Download presentation
Presentation is loading. Please wait.

1
RBC and BLEEDING DISORDERS

2
RBC and Bleeding Disorders NORMAL NORMAL – Anatomy, histology – Development – Physiology ANEMIAS ANEMIAS – Blood loss: acute, chronic – Hemolytic – Diminished erythropoesis POLYCYTHEMIA POLYCYTHEMIA BLEEDING DISORDERS BLEEDING DISORDERS

6
TABLE 13-2 -- Adult Reference Ranges for Red Blood Cells ** Measurement (units)MenWomen Hemoglobin (gm/dL)13.6–17.212.0–15.0 Hematocrit (%)39–4933–43 Red cell count (10 6 /µL)4.3–5.93.5–5.0 Reticulocyte count (%)0.5–1.5 Mean cell volume (µm 3 ) MCV82–96 Mean corpuscular hemoglobin (pg) MCH27–33 Mean corpuscular hemoglobin concentration (gm/dL) MCHC 33–37 RBC distribution width11.5–14.5
MenWomen Hemoglobin (gm/dL)13.6– –15.0 Hematocrit (%)39–4933–43 Red cell count (10 6 /µL)4.3–5.93.5–5.0 Reticulocyte count (%)0.5–1.5 Mean cell volume (µm 3 ) MCV82–96 Mean corpuscular hemoglobin (pg) MCH27–33 Mean corpuscular hemoglobin concentration (gm/dL) MCHC 33–37 RBC distribution width11.5–14.5")
8
WHERE is MARROW? Yolk Sac: very early embryo Liver, Spleen: NEWBORN BONE – CHILDHOOD: AXIAL SKELETON & APPENDICULAR SKELETON BOTH HAVE RED (active) MARROW – ADULT: AXIAL SKELETON RED MARROW, APPENDICULAR SKELETON YELLOW MARROW
 MARROW – ADULT: AXIAL SKELETON RED MARROW, APPENDICULAR SKELETON YELLOW MARROW.")
9
MARROW FEATURES CELLULARITY 50% MEGAKARYOCYTES at least 1-2/hpf M:E RATIO 3:1 MYELOID MATURATION 1/3 bands or more ERYTHROID MATURATION nucleus/cytoplasm LYMPHS, PLASMA CELLS small percentage STORAGE IRON, i.e., HEMOSIDERIN present “FOREIGN CELLS”

10
MARROW “DIFFERENTIATION”

12
ANEMIAS* BLOOD LOSS – ACUTE – CHRONIC IN-creased destruction (HEMOLYTIC) DE-creased production * A good definition would be a decrease in OXYGEN CARRYING CAPACITY, rather than just a decrease in red blood cells, because you need to have enough blood cells THAT FUNCTION, and not just enough blood cells.
 DE-creased production * A good definition would be a decrease in OXYGEN CARRYING CAPACITY, rather than just a decrease in red blood cells, because you need to have enough blood cells THAT FUNCTION, and not just enough blood cells.")
13
Features of ALL anemias Pallor, where? Tiredness Weakness Dyspnea, why? Palpitations Heart Failure (high output), why?
, why .")
14
Blood Loss Acute: trauma Chronic: lesions of gastrointestinal tract, gynecologic disturbances. The features of chronic blood loss anemia are the same as iron deficiency anemia, and is defined as a situation in which the production cannot keep up with the loss.

15
HEMOLYTIC HEREDITARY – MEMBRANE disorders: e.g., spherocytosis – ENZYME disorders: e.g., G6PD deficciency – HGB disorders (hemoglobinopathies) ACQUIRED – MEMBRANE disorders (PNH) – ANTIBODY MEDIATED, transfusion or autoantibodies – MECHANICAL TRAUMA (vascular or mechanic) – INFECTIONS – DRUGS, TOXINS – HYPERSPLENISM
 ACQUIRED – MEMBRANE disorders (PNH) – ANTIBODY MEDIATED, transfusion or autoantibodies – MECHANICAL TRAUMA (vascular or mechanic) – INFECTIONS – DRUGS, TOXINS – HYPERSPLENISM")
16
IMPAIRED PRODUCTION Disturbance of proliferation and differentiation of stem cells: aplastic anemias, pure RBC aplasia, renal failure Disturbance of proliferation and maturation of erythroblasts Defective DNA synthesis: (Megaloblastic) Defective heme synthesis: (Fe) Deficient globin synthesis: (Thalassemias)
 Defective heme synthesis: (Fe) Deficient globin synthesis: (Thalassemias)")
17
MODIFIERS MCV, microcytosis, macrocytosis MCH MCHC, hypochromic RDW, anisocytosis

18
HEMOLYTIC ANEMIAS Life span LESS than 120 days Marrow hyperplasia (M:E), EPO+ Increased catabolic products, e.g., bilirubin, serum HGB, hemosiderin, haptoglobin-HGB
, EPO+ Increased catabolic products, e.g., bilirubin, serum HGB, hemosiderin, haptoglobin-HGB")
19
HEMOLYSIS INTRA-vascular (vessels) EXTRA-vascular (spleen)
 EXTRA-vascular (spleen)")
20
M:E Ratio normally 3:1

21
HEREDITARY SPHEROCYTOSIS Genetic defects affecting ankyrin, spectrin, usually autosomal dominant Children, adults Anemia, hemolysis, jaundice, splenomegaly, gallstones (what kind?)
")
22
Glucose-6-Phosphate Dehydrogenase (G6PD) Deficiency A - and Mediterranean are most significant types
 Deficiency A - and Mediterranean are most significant types")
23
FEATURES of G6PD Defic. Genetic: X-linked Can be triggered by foods (fava beans), oxidant substances drugs (primaquine, chloroquine), or infections HGB can precipitate as HEINZ bodies Acute intravascular hemolysis can occur: – Hemoglobinuria – Hemoglobinemia – Anemia
, oxidant substances drugs (primaquine, chloroquine), or infections HGB can precipitate as HEINZ bodies Acute intravascular hemolysis can occur: – Hemoglobinuria – Hemoglobinemia – Anemia.")
24
Sickle Cell Disease Classic hemoglobinopathy Normal HGB is α2 β2: β-chain defects (Val->Glu) Reduced hemoglobin “sickles” in homozygous 8% of American blacks are heterozygous
 Reduced hemoglobin sickles in homozygous 8% of American blacks are heterozygous")
25
Clinical features of HGB-S disease Severe anemia Jaundice PAIN (pain CRISIS) Vaso-occlusive disease: EVERYWHERE, but clinically significant bone, spleen (autosplenectomy) Infections: Pneumococcus, Hem. Influ., Salmonella osteomyelitis

27
THALASSEMIAS A WIDE VARIETY of diseases involving GLOBIN synthesis, COMPLEX genetics Alpha or beta chains deficient synthesis involved Often termed MAJOR or MINOR, depending on severity, silent carriers and “traits” are seen HEMOLYSIS is uniformly a feature, and microcytic anemia, i.e, LOW MCV (just like iron deficiency anemia has a low MCV) A “crew cut” skull x-ray appearance may be seen in severe erythroid hyperplasia.
 A crew cut skull x-ray appearance may be seen in severe erythroid hyperplasia.")
29
Hemoglobin H Disease Deletion of THREE alpha chain genes HGB-H is primarilly Asian HGB- H has a HIGH affinity for oxygen HGB-H is unstable and therefore has classical hemolytic behavior

30
HYDROPS FETALIS FOUR alpha chain genes are deleted, so this is the MOST SEVERE form of thalassemia Many/most never make it to term Children born will have a SEVERE hemolytic anemia as in the erythroblastosis fetalis of Rh disease: – Pallor (as in all anemias), jaundice, kernicterus – Edema (hence the name “hydrops”) – Massive hepatosplenomegaly (hemolysis)
, jaundice, kernicterus – Edema (hence the name hydrops ) – Massive hepatosplenomegaly (hemolysis)")
31
Paroxysmal Nocturnal Hemoglobinuria (PNH) ACQUIRED, NOT INHERITED like all the previous hemolytic anemias were ACQUIRED mutations in phosphatidylinositol glycan A (PIGA) Note: It is “P” and “N” only 25% of the time! G lycosylphos P hatidyl I nositol (lipid rafts)
.")
32
Immunohemolytic Anemia All of these have the presence of antibodies and/or compliment present on RBC surfaces NOT all are AUTOimmune, some are caused by drugs Antibodies can be – WARM AGGLUTININ (IgG) – COLD AGGLUTININ (IgM) – COLD HEMOLYSIN (paroxysmal) (IgG)
 – COLD AGGLUTININ (IgM) – COLD HEMOLYSIN (paroxysmal) (IgG)")
33
IMMUNOHEMOLYTIC ANEMIAS WARM AGGLUTININS (IgG), will NOT hemolyze at room temp – Primary Idiopathic (most common) – Secondary (Tumors, especially leuk/lymph, drugs) COLD AGGLUTININS: (IgM), WILL hemolyze at room temp – Mycoplasma pneumoniae, HIV, mononucleosis COLD HEMOLYSINS: (IgG) Cold Paroxysmal Hemoglobinuria, hemo-LYSIS in body, ALSO often follows mycoplasma pneumoniae
, will NOT hemolyze at room temp – Primary Idiopathic (most common) – Secondary (Tumors, especially leuk/lymph, drugs) COLD AGGLUTININS: (IgM), WILL hemolyze at room temp – Mycoplasma pneumoniae, HIV, mononucleosis COLD HEMOLYSINS: (IgG) Cold Paroxysmal Hemoglobinuria, hemo-LYSIS in body, ALSO often follows mycoplasma pneumoniae")
34
COOMBS TEST DIRECT: Patient’s CELLS are tested for surface Ab’s INDIRECT: Patient’s SERUM is tested for Ab’s.

35
HEMOLYSIS/HEMOLYTIC ANEMIAS DUE TO RBC TRAUMA Mechanical heart valves breaking RBC’s MICROANGIOPATHIES: – TTP – Hemolytic Uremic Syndrome

36
NON-Hemolytic Anemias: i.e., DE-creased Production “Megaloblastic” Anemias B12 Deficiency (Pernicious Anemia) Folate Deficiency Iron Deficiency Anemia of Chronic Disease Aplastic Anemia “Pure” Red Cell Aplasia OTHER forms of Marrow Failure
 Folate Deficiency Iron Deficiency Anemia of Chronic Disease Aplastic Anemia Pure Red Cell Aplasia OTHER forms of Marrow Failure")
37
MEGALOBLASTIC ANEMIAS Differentiating megaloblasts (marrow) from macrocytes (peripheral smear, MCV>94) Impaired DNA synthesis For all practical purposes, also called the anemias of B12 and FOLATE deficiency Often VERY hyperplastic/hypercellular marrow* (* exception to the rule)
 from macrocytes (peripheral smear, MCV>94) Impaired DNA synthesis For all practical purposes, also called the anemias of B12 and FOLATE deficiency Often VERY hyperplastic/hypercellular marrow* (* exception to the rule)")
38
Decreased intake Inadequate diet, vegetarianism Impaired absorption Intrinsic factor deficiency Pernicious anemia Gastrectomy Malabsorption states Diffuse intestinal disease, e.g., lymphoma, systemic sclerosis Ileal resection, ileitis Competitive parasitic uptake Fish tapeworm infestation Bacterial overgrowth in blind loops and diverticula of bowel Increased requirement Pregnancy, hyperthyroidism, disseminated cancer

39
Vit-B12 Physiology Oral ingestion Combines with INTRINSIC FACTOR in the gastric mucosa Absorbed in the terminal ileum DEFECTS at ANY of these sites can produce a MEGALOBLASTIC anemia

40
Please remember that ALL megaloblastic anemias are also MACROCYTIC (MCV>94 or MCV~100), and that not only are the RBC’s BIG and hyperplastic/hypercellular, but so are the neutrophils, and neutrophilic precursors in the bone marrow too, and even more so, HYPERSEGMENTED!!!
, and that not only are the RBC’s BIG and hyperplastic/hypercellular, but so are the neutrophils, and neutrophilic precursors in the bone marrow too, and even more so, HYPERSEGMENTED!!!")
41
PERNICIOUS ANEMIA MEGALOBLASTIC anemia LEUKOPENIA and HYPERSEGS JAUNDICE… ??? NEUROLOGIC posterolateral spinal tracts ACHLORHYDRIA Can’t absorb B12 LOW serum B12 Flunk Schilling test, i.e., can’t absorb B12, using a radioactive tracer

42
FOLATE DEFICIENCY MEGALOBLASTIC AMEMIAS Decreased Intake: diet, etoh-ism, infancy Impaired Absorption: intestinal disease DRUGS: anticonvulsants, BCPs, CHEMO Increased Loss: hemodialysis Increased Requirement: pregnancy, infancy Impaired Usage

43
Fe Deficiency Anemia Due to increased loss or decreased ingestion, almost always, in USA, nowadays, increased loss is the reason Microcytic (low MCV), Hypochromic (low MCHC) THE ONLY WAY WE CAN LOSE IRON IS BY LOSING BLOOD, because FE is recycled!
, Hypochromic (low MCHC) THE ONLY WAY WE CAN LOSE IRON IS BY LOSING BLOOD, because FE is recycled!")
44
Fe Transferrin Ferritin/apo- (GREAT test) Hemosiderin
 Hemosiderin")
45
Clinical Fe-Defic-Anemia Adult men: GI Blood Loss PRE menopausal women: menorrhagia POST menopausal women: GI Blood Loss

47
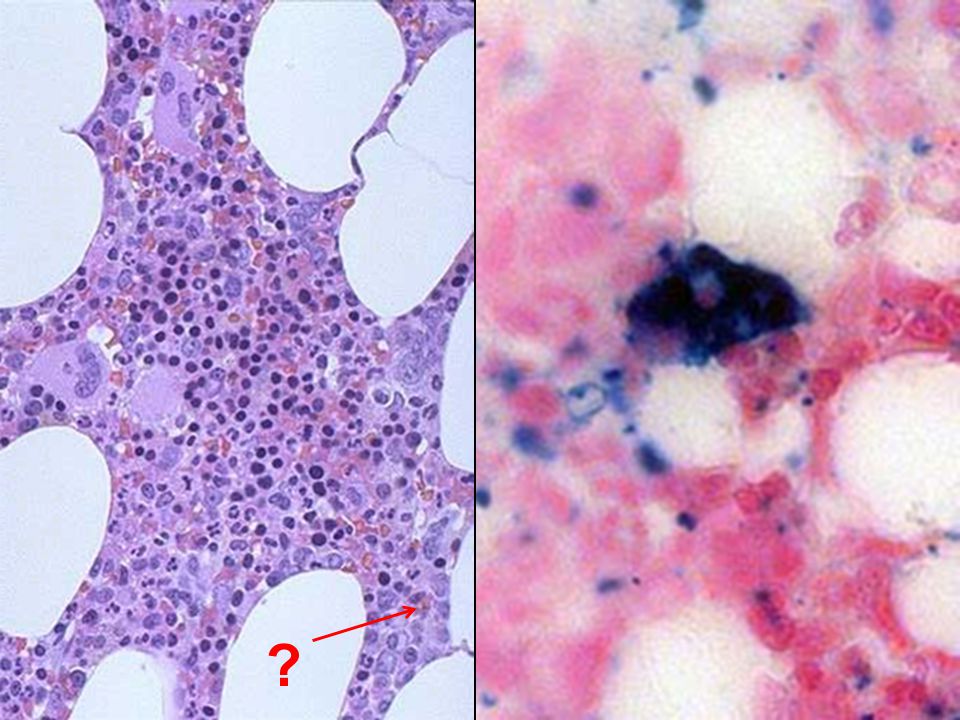
2 BEST lab tests: Serum Ferritin Prussian blue hemosiderin stain of marrow (also called an “iron” stain)
")
48
?
49
Anemia of Chronic Disease* CHRONIC INFECTIONS CHRONIC IMMUNE DISORDERS NEOPLASMS LIVER, KIDNEY failure * Please remember these patients may very very much look like iron deficiency anemia, BUT, they have ABUNDANT STAINABLE HEMOSIDERIN in the marrow!

50
APLASTIC ANEMIAS ALMOST ALWAYS involve platelet and WBC suppression as well Some are idiopathic, but MOST are related to drugs, radiation FANCONI’s ANEMIA is the only one that is inherited, and NOT acquired Act at STEM CELL level, except for “pure” red cell aplasia

51
APLASTIC ANEMIAS

52
CHLORAMPHENICOL OTHER ANTIBIOTICS CHEMO INSECTICIDES, benzene, toluene, TNT VIRUSES – EBV – HEPATITIS – VZ

53
MYELOPHTHISIC ANEMIAS Are anemias caused by metastatic tumor cells replacing the bone marrow extensively

54
POLYCYTHEMIA Relative (e.g., hemoconcentration) Absolute – POLYCYTHEMIA VERA (Primary) ( LOW EPO), mutation in tyrosine kinase, making RBCs hyper responsive to EPO – POLYCYTHEMIA (Secondary) (HIGH EPO) HIGH ALTITUDE EPO TUMORS EPO “Doping” CVAC, the trendy California bubble pods
 Absolute – POLYCYTHEMIA VERA (Primary) ( LOW EPO), mutation in tyrosine kinase, making RBCs hyper responsive to EPO – POLYCYTHEMIA (Secondary) (HIGH EPO) HIGH ALTITUDE EPO TUMORS EPO Doping CVAC, the trendy California bubble pods")
55
P. VERA A “myeloproliferative” disease ALL cell lines are increased, not just RBCs

56
BLEEDING DISORDERS (aka, Hemorrhagic “DIATHESES”) Blood vessel wall abnormalities √ Reduced platelets √ Decreased platelet function √ Abnormal clotting factors √ DIC ( D isseminated I NTRA-vascular C oagulation), also has ↓ plats.
 Blood vessel wall abnormalities √ Reduced platelets √ Decreased platelet function √ Abnormal clotting factors √ DIC ( D isseminated I NTRA-vascular C oagulation), also has ↓ plats.")
57
VESSEL WALL ABNORMALITIES (angiopathic thrombocytopenias) (NON-thrombotic purpuras) Infections, especially, meningococcemia, and rickettsia Drug reactions causing a leukocytoclastic vasculitis Scurvy, Ehlers-Danlos, Cushing syndrome Henoch-Schönlein purpura (mesangial deposits too) Hereditary hemorrhagic telangiectasia Amyloid
 (NON-thrombotic purpuras) Infections, especially, meningococcemia, and rickettsia Drug reactions causing a leukocytoclastic vasculitis Scurvy, Ehlers-Danlos, Cushing syndrome Henoch-Schönlein purpura (mesangial deposits too) Hereditary hemorrhagic telangiectasia Amyloid")
58
THROMBOCYTOPENIAS Like RBCs: – DE-creased production – IN-creased destruction – Sequestration (Hypersplenism) – Dilutional Normal value 150K-300K
 – Dilutional Normal value 150K-300K")
59
DE-CREASED PRODUCTION APLASTIC ANEMIA ACUTE LEUKEMIAS ALCOHOL, THIAZIDES, CHEMO MEASLES, HIV MEGALOBLASTIC ANEMIAS MYELODYSPLASTIC SYNDROMES

60
IN-CREASED DESTRUCTION AUTOIMMUNE (ITP) POST-TRANSFUSION (NEONATAL) QUINIDINE, HEPARIN, SULFA MONO, HIV DIC TTP “MICROANGIOPATHIC”
 POST-TRANSFUSION (NEONATAL) QUINIDINE, HEPARIN, SULFA MONO, HIV DIC TTP MICROANGIOPATHIC")
61
THROMBOCYTOPENIAS ITP (Idiopathic Thrombocytopenic Purpura) Acute Immune DRUG-induced HIV associated TTP, Hemolytic Uremic Syndrome
 Acute Immune DRUG-induced HIV associated TTP, Hemolytic Uremic Syndrome")
62
I.T.P. ADULTS AND ELDERLY ACUTE OR CHRONIC AUTO-IMMUNE ANTI-PLATELET ANTIBODIES PRESENT INCREASED MARROW MEGAKARYOCYTES Rx: STEROIDS

63
ACUTE ITP CHILDREN Follows a VIRAL illness (~ 2 weeks) ALSO have anti-platelet antibodies Platelets usually return to normal in a few months
 ALSO have anti-platelet antibodies Platelets usually return to normal in a few months")
64
DRUGS Quinine Quinidine Sulfonamide antibiotics HEPARIN

65
HIV BOTH DE-creased production AND IN-creased destruction factors are present

66
Thrombotic Microangiopathies BOTH are very SERIOUS CONDITIONS with a HIGH mortality: – TTP (THROMBOTIC THROMBOCYTOPENIC PURPURA) – H.U.S. (HEMOLYTIC UREMIC SYNDROME) These can also be called “consumptive” coagulopathies, just like a DIC
 These can also be called consumptive coagulopathies, just like a DIC.")
67
“QUALITATIVE” platelet disorders Mostly congenital (genetic): – Bernard-Soulier syndrome (Glycoprotein-1- b deficiency) – Glanzmann’s thrombasthenia (Glyc.-IIB/IIIA deficiency) – Storage pool disorders, i.e., platelets mis- function because of defective granulation ACQUIRED: ASPIRIN, ASPIRIN, ASPIRIN
: – Bernard-Soulier syndrome (Glycoprotein-1- b deficiency) – Glanzmann’s thrombasthenia (Glyc.-IIB/IIIA deficiency) – Storage pool disorders, i.e., platelets mis- function because of defective granulation ACQUIRED: ASPIRIN, ASPIRIN, ASPIRIN")
68
PTT PT/INR

69
BLEEDING DISORDERS due to CLOTTING FACTOR DEFICIENCIES NOT spontaneous, but following surgery or trauma ALL factor deficiencies are possible Factor VIII and IX both are the classic X-linked recessive hemophilias, A and B, respectively ACQUIRED disorders often due to Vitamin-K deficiencies (II, VII, IX, X) von Willebrand disease the most common, 1%
 von Willebrand disease the most common, 1%")
70
von Willebrand Disease 1% prevalence, most common bleeding disorder Spontaneous and wound bleeding Usually autosomal dominant Gazillions of variants, genetics even more complex Prolonged BLEEDING TIME, NL platelet count vWF is von Willebrand Factor, which complexes with Factor VIII, to join platelets with the exposed ECM in endothelial disruption. it is the von Willebrand Factor which is defective in von Willebrand disease Usually BOTH platelet and FactorVIII-vWF disorders are present

71
PTT PT/INR

72
HEMOPHILIA A The “classic” HEMOPHILIA Factor VIII decreased Co-factor of Factor IX to activate Factor X Sex-linked recessive Hemorrhage usually NOT spontaneous Wide variety of severities Prolonged PTT (intrinsic) only Rx: Recombinant Factor VIII
 only Rx: Recombinant Factor VIII")
73
HEMOPHILIA B The “Christmas” HEMOPHILIA Factor IX decreased Sex-linked recessive Hemorrhage usually NOT spontaneous Wide variety of severities Prolonged PTT (intrinsic) only Rx: Recombinant Factor IX
 only Rx: Recombinant Factor IX")
74
DIC, Disseminated INTRA-vascular, Coagulation ENDOTHELIAL INJURY WIDESPREAD FIBRIN DEPOSITION HIGH MORTALITY ALL MAJOR ORGANS COMMONLY INVOLVED

75
DIC, Disseminated INTRA-vascular, Coagulation Extremely SERIOUS condition NOT a disease in itself but secondary to many conditions – Obstetric: MAJOR OB complications, toxemia, sepsis, abruption – Infections: Gm-, meningococcemia, RMSF, fungi, Malaria – Many neoplasms, acute promyelocytic leukemia – Massive tissue injury: trauma, burns, surgery “Consumptive” coagulopathy

76
Common Coagulation TESTS PTT (intrinsic) PT INR (extrinsic) Platelet count, aggregation Bleeding Time, so EASY to do Fibrinogen Factor Assays
 PT INR (extrinsic) Platelet count, aggregation Bleeding Time, so EASY to do Fibrinogen Factor Assays")
77
RBC LAB http://www.chronolab.com/hematology/2_1.htm

Similar presentations















. Complete Blood Count ( CBC)>")










