Download presentation
Presentation is loading. Please wait.

1
Case Report # [] Submitted by:Kandra Vogt, MSIV Faculty reviewer:Sandra A. A. Oldham, M.D. Date accepted:31 August 2007 Radiological Category:Principal Modality (1): Principal Modality (2): Genitourinary Ultrasound CT
![Case Report # [] Submitted by:Kandra Vogt, MSIV Faculty reviewer:Sandra A.](http://images.slideplayer.com/18/5671912/slides/slide_1.jpg "A. Oldham, M.D. Date accepted:31 August 2007 Radiological Category:Principal Modality (1): Principal Modality (2): Genitourinary Ultrasound CT.")
2
Case History 43 yo WF who presents with HTN and hematuria

3
Radiological Presentations

6
at Age 71

7
von Hippel-Lindau disease Autosomal recessive polycystic kidney disease Multiple simple cysts Autosomal dominant polycystic kidney disease Acquired cystic kidney disease Tuberous sclerosis Which one of the following is your choice for the appropriate diagnosis? After your selection, go to next page. Test Your Diagnosis

8
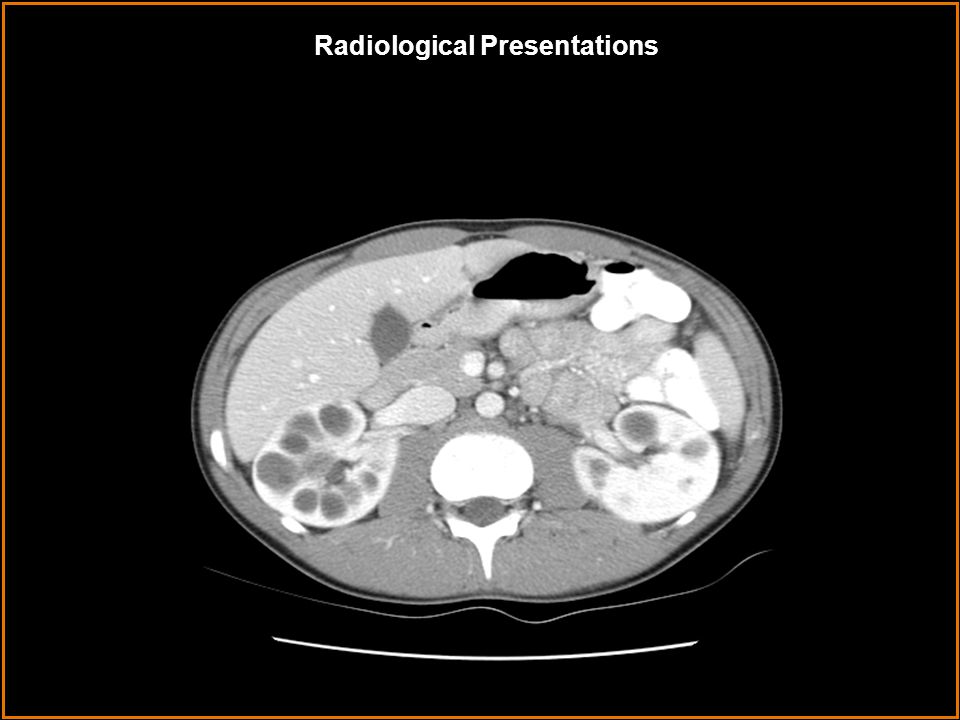
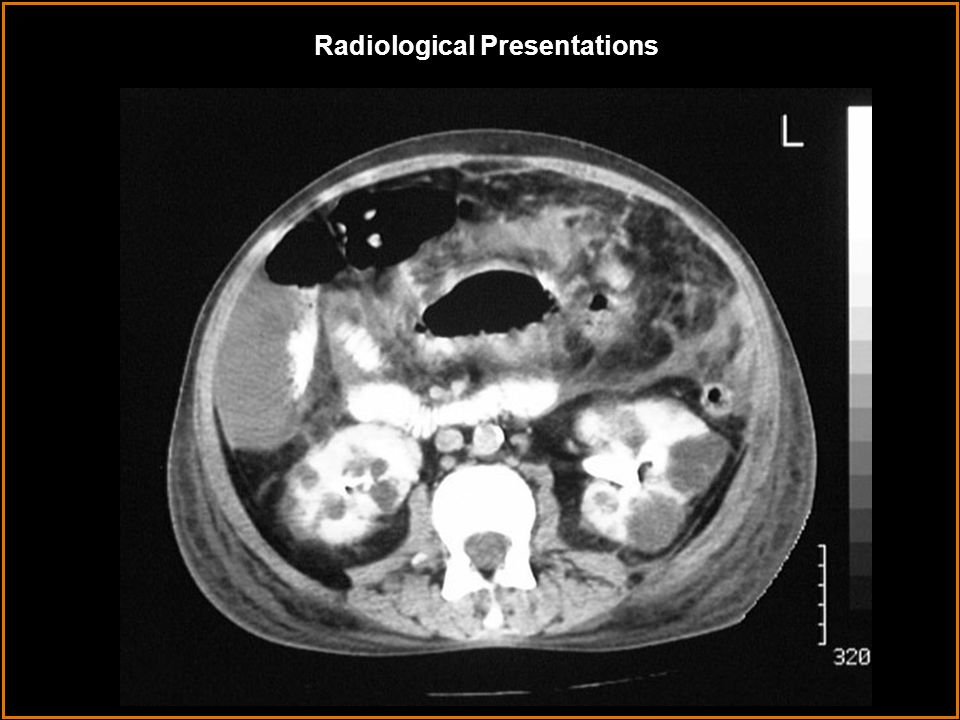
Image 2 shows bilaterally enlarged kidneys with well-defined, multifocal round lesions of varying size with low attenuation values similar to water. After the administration of IV contrast, cysts do not enhance but stand out prominently against normally enhancing background renal tissue. Curvilinear pattern of calcification is also common, although not seen here. Image 4 shows a different patient with long standing polycystic kidney disease s/p bilateral nephrectomies and renal transplant. There are multiple varying in size well- defined hypodensities within the liver involving both lobes. Definitive evaluation without IV contrast cannot be made; however, most likely, these represent cysts. There are surgical clips in both renal fossa indicating previous bilateral nephrectomies. Autosomal Dominant PKD Multiple simple renal cysts Acquired cystic kidney disease Autosomal Recessive PKD von Hippel-Lindau disease Tuberous sclerosis Findings: Differentials: Findings and Differentials

9
Autosomal Dominant Polycystic Kidney Disease (ADPKD) is a slowly progressive disease with nearly 100% penetrance and great variation in expressivity. One of every 1000 people carry the mutant gene but spontaneous mutations occur as well. The gene is located on chromosome 16p in 90% of cases, the PKD1 locus. Defective polycystins appear to contribute to cyst formation by affecting epithelial cell maturation, resulting in the development of cysts of varying sizes in the cortex and medulla. It is the most common hereditary kidney disease and 3 rd most prevalent cause of chronic renal failure. Clinical manifestations include HTN, hematuria, renal failure and progressive RI, or abdominal pain. The mean age at diagnosis is 43 years but patients present commonly between the 3 rd and 5 th decade of life. The M:F ratio is 1:1. The onset of cyst formation is apparent in 54% by the 1 st decade of life, 72% by the 2 nd decade, 80% by the 3 rd decade, and all by age 80. Discussion

10
Pathophysiology: The renal parenchyma is progressively replaced with non-communicating cysts. As the cysts increase in number and size, the kidneys enlarge asymmetrically and lose their normal shape. Diagnosis is made by genetic studies and ultrasound or CT combined with the patient’s clinical history. Clinical Course: By age 60, most with ADPKD experience renal failure. Histologic exam of tissue suggests that the progressive renal failure correlates most closely with the development of vascular sclerosis and interstitial fibrosis. How these changes occur and their possible relationship to cyst formation are not clear. They are most commonly treated with hemodialysis or renal transplantation and often require nephrectomy. Discussion

11
Extrarenal manifestations: ADPKD is associated with cysts in the liver (25-50%), pancreas (9%), and are rare in the lung, spleen, thyroid, ovaries, uterus, testis, seminal vesicles, epididymis, and bladder. Saccular “berry” aneurysms in the Circle of Willis are found in 10-30% of patients so screening with MRA is recommended. A variety of cardiac and aortic abnormalities have been associated with ADPKD, including aortic root dilatation, aortic regurgitation, bicuspid aortic valves, coarctation of the aorta, mitral regurgitation, and abdominal aortic aneurysm. Complications include death from uremia (59%), cerebral hemorrhage (13%), infection, or cardiac complications, renal calculi, urinary tract infections, cyst rupture, hemorrhage, and infection. The incidence of renal cell carcinoma is only slightly increased in patients with ADPKD. Discussion
, cerebral hemorrhage (13%), infection, or cardiac complications, renal calculi, urinary tract infections, cyst rupture, hemorrhage, and infection. The incidence of renal cell carcinoma is only slightly increased in patients with ADPKD. Discussion.")
12
Cerebral aneurysms in the Circle of Willis

13
Multiple simple renal cysts: Patients are usually older, have fewer cysts, usually no renal failure, and no family history of cystic kidney disease. There are no cysts in other organs. Tuberous sclerosis: This condition has an autosomal dominant inheritance pattern. Manifestations of tuberous sclerosis can become apparent in persons of any age, but most patients have clinical symptoms before they age 10. The classic form of the disease is the triad of seizures, mental retardation, and adenoma sebaceum. Patients have combined multiple angiomyolipomas and multiple renal cysts associated with cutaneous, retinal, and cerebral hamartomas. von Hippel-Lindau disease: These patients often present in the 2 nd to 3 rd decade of life. There are multiple renal and pancreatic cysts with the addition of cerebellar hemangioblastomas, pheochromocytomas, retinal hemangiomas +/- bilateral renal cell carcinomas. CNS hemangioblastoma (Lindau tumor) is the most commonly recognized manifestation of VHL and occurs in 40% of patients. Discussion
 is the most commonly recognized manifestation of VHL and occurs in 40% of patients. Discussion.")
14
Acquired cystic kidney disease: Develops in patients requiring long-term dialysis. The kidneys are usually small, reflecting chronic renal disease. The cysts rarely exceed 2 cm. A utosomal Recessive PKD: This disease primarily occurs in infants and children and the cysts are usually microscopic. Liver disease is present in every case and is described as periportal fibrosis and biliary duct ectasia. Consequently, patients develop portal hypertension. Discussion

15
Autosomal dominant polycystic kidney disease Diagnosis

16
Dahnert W. Radiology Review Manual. 4 th ed. Philadelphia, PA: Williams and Wilkins, 1999:782-783 Brant WE, Helms CA. Fundamentals of Diagnostic Radiology. 2 nd ed. Philadelphia, PA: Williams and Wilkins, 1999:782-784 Chapman AB, Rahbari FF, Bennett WM. Course and treatment of autosomal dominant polycystic kidney disease. 23 March 2007. Up To Date. Rose BD, Bennett WM. Diagnosis of and screening for autosomal dominant polycystic kidney disease. 22 January 2007. Up To Date. Khan AN. Autosomal Dominant Polycystic Kidney Disease. 12 January 2007. www.emedicine.com References

Similar presentations












 tumors arising from one of the many different cell types within.>")

: Principal Modality (2): Neuroradiology.>")




: Principal Modality (2): PET/CT CT Faculty Reviewer:>")