Download presentation
Presentation is loading. Please wait.

1
Sickle cell anemia Clinical vignettes
Peter Newburger, MD Pediatric hematology/oncology

2
Sickle cell anemia From

3
Sickle Cell Epidemiology
bS : Single a.a. substitution Glutamic acid Valine Most common single gene disorder in African Americans 1/375 affected (homozygous) 1/12 are heterozygous carriers (~8%) Also affects other ethnicities: India, Middle East, Hispanic
 1/12 are heterozygous carriers (~8%) Also affects other ethnicities: India, Middle East, Hispanic.")
4
SICKLE CELL SYNDROMES Molecular pathology postulated by Pauling in the late 1940s Mutation known for 50 years Unfinished tasks Explaining the clinical disorder based on the molecular defect Rational and targeted treatments – still few despite detailed knowledge of the molecular defect

5
Molecular Pathophysiology
Intracellular [Hb] ~ g/dL Deoxygenation allows interaction of bS subunits via abnormal hydrophobic regions (valines) Non-covalent bond with other bS in the RBC Formation of 14 stranded helical fiber
 Non-covalent bond with other bS in the RBC. Formation of 14 stranded helical fiber.")
6
Delay time ≈ k / C15 The time that elapses between the deoxygenation of hemoglobin S and the formation of polymer is inversely proportional to the intracellular concentration (C) of deoxyhemoglobin, raised to the 15th power
 of deoxyhemoglobin, raised to the 15th power.")
7
Polymerization phase is sensitive to: O2 concentration
Hgb concentration pH Ionic strength (At salt concentrations spanning the physiologic range, solubility increases with ionic strength, but decreases markedly at high ionic strength.) Induction of Red-Cell Sickling by Polymerization of Deoxyhemoglobin S. As red cells traverse the microcirculation, oxygen is released from oxyhemoglobin (red circles), generating deoxyhemoglobin (purple circles). The diagram at the left of the figure shows molecules of hemoglobin S, with the globular 2 S2 tetramer shown as a flat circle. Deoxygenation of hemoglobin S induces a change in conformation in which the subunits move away from each other. The hydrophobic patch at the site of the 6 valine replacement, shown as a projection, can bind to a complementary hydrophobic site on a subunit of another hemoglobin tetramer, shown as an indentation. This interaction is necessary for the formation of polymer, depicted as the interaction of three deoxyhemoglobin S molecules on one strand (dark purple) with three deoxyhemoglobin S molecules on another strand (light purple). At the bottom, a high-resolution model, prepared by Drs. Robert Josephs, S.J. Watowich, and L.J. Gross, shows the interaction of three deoxyhemoglobin S molecules on one strand with three deoxyhemoglobin S molecules on another strand. The subunits are pale yellow-green, and the subunits are gray (lighter in the foreground and darker in the background). The heme groups are shown as red spheres. Also shown are contacts between foreground subunits involving 6 valine (blue) on one strand and the hydrophobic acceptor site (bright green) on the other strand. Only one of the two 6 valine sites in each hemoglobin S tetramer makes this contact. The diagram in the middle shows the assembly of deoxyhemoglobin S into a helical 14-strand fiber, shown as a twisted rope-like structure. The equation shows the time that elapses, or delay time (td), between the deoxygenation of hemoglobin S and the concerted formation of polymer. The delay time is inversely proportional to the intracellular hemoglobin concentration (C), raised to about the 15th power; k denotes an experimental constant. The photograph at the bottom is a high-resolution electron micrograph of a fiber, provided by Dr. Stuart Edelstein. As deoxyhemoglobin S polymerizes and fibers align, the red cell is distorted into an elongated banana or "sickle" shape, as shown in the diagram at the right. The photograph at the bottom is a scanning electron micrograph of a reversibly sickled cell, provided by Dr. James White. Bunn HF. NEJM (11)
 Induction of Red-Cell Sickling by Polymerization of Deoxyhemoglobin S. As red cells traverse the microcirculation, oxygen is released from oxyhemoglobin (red circles), generating deoxyhemoglobin (purple circles). The diagram at the left of the figure shows molecules of hemoglobin S, with the globular 2 S2 tetramer shown as a flat circle. Deoxygenation of hemoglobin S induces a change in conformation in which the subunits move away from each other. The hydrophobic patch at the site of the 6 valine replacement, shown as a projection, can bind to a complementary hydrophobic site on a subunit of another hemoglobin tetramer, shown as an indentation. This interaction is necessary for the formation of polymer, depicted as the interaction of three deoxyhemoglobin S molecules on one strand (dark purple) with three deoxyhemoglobin S molecules on another strand (light purple). At the bottom, a high-resolution model, prepared by Drs. Robert Josephs, S.J. Watowich, and L.J. Gross, shows the interaction of three deoxyhemoglobin S molecules on one strand with three deoxyhemoglobin S molecules on another strand. The subunits are pale yellow-green, and the subunits are gray (lighter in the foreground and darker in the background). The heme groups are shown as red spheres. Also shown are contacts between foreground subunits involving 6 valine (blue) on one strand and the hydrophobic acceptor site (bright green) on the other strand. Only one of the two 6 valine sites in each hemoglobin S tetramer makes this contact. The diagram in the middle shows the assembly of deoxyhemoglobin S into a helical 14-strand fiber, shown as a twisted rope-like structure. The equation shows the time that elapses, or delay time (td), between the deoxygenation of hemoglobin S and the concerted formation of polymer. The delay time is inversely proportional to the intracellular hemoglobin concentration (C), raised to about the 15th power; k denotes an experimental constant. The photograph at the bottom is a high-resolution electron micrograph of a fiber, provided by Dr. Stuart Edelstein. As deoxyhemoglobin S polymerizes and fibers align, the red cell is distorted into an elongated banana or sickle shape, as shown in the diagram at the right. The photograph at the bottom is a scanning electron micrograph of a reversibly sickled cell, provided by Dr. James White. Bunn HF. NEJM (11)")
9
Cellular Pathophysiology of Sickle Cell syndromes
Polymerization leads to: Distortion of Cell shape Damage to RBC Membrane Abnormal permeability Irreversible sickling Impairment of RBC flow = Infarction Decreased red cell number = Anemia

10
Anemia Most common feature of sickle cell disease
Often ignored as pathologic Moderate to severe in almost all patients Degree of anemia reflects clinical severity Episodic acute anemia Anemia in Sickle Cell Disease per se is not viewed as a complication that requires treatment, but anemia has been associated with some of the major complications of the disease.

11
Sickle complications Vaso-occlusive crisis Cerebrovascular disease
Splenic sequestration Sepsis due to functional asplenia Acute chest syndrome

12
Patient vignettes

13
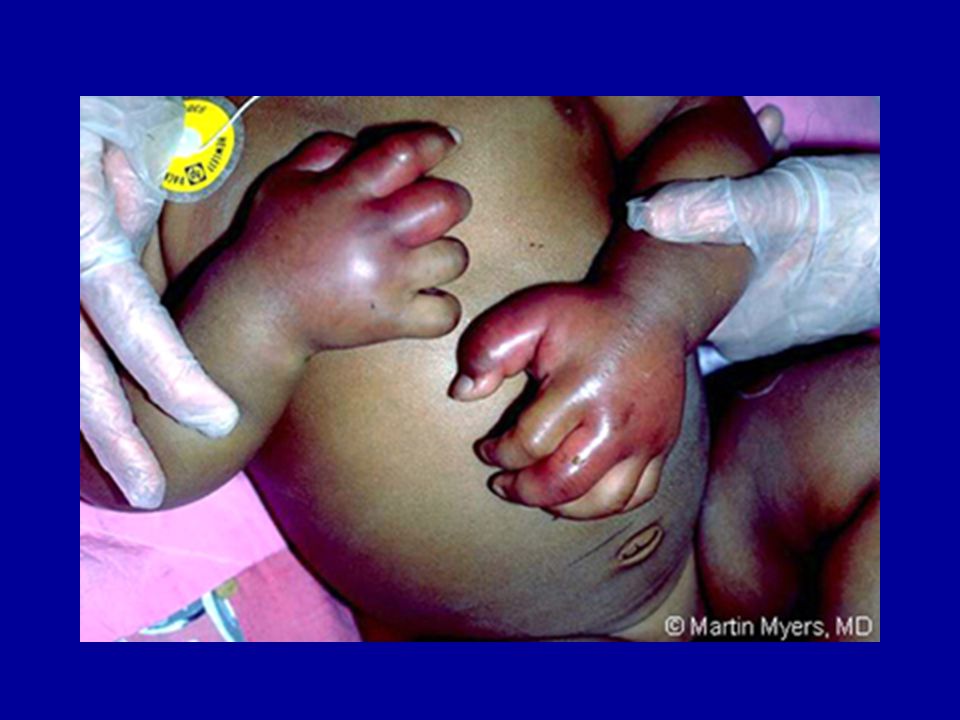
18 month old girl presents to the ER crying inconsolably, with fever and swollen, tender hands.

15
A 3 year old boy presents to the ER with a 12-hour history of fever to 38°C. He is slightly irritable but looks well. Despite IV antiobiotics, his fever continues to rise, his blood pressure falls, and his extremities become cold, with purple discoloration.

17
Functional asplenia Functional Asplenia
Liver spleen scans from two children with HbSS. The scan at left shows functioning splenic tissue in the left upper quadrant along with normal hepatic uptake. The scan on the right shows normal hepatic uptake but absence of splenic function resulting from repeated splenic infarction. Functional Asplenia

18
Sepsis Prevention: the most effective drug for sickle cell is…
Volume 314: June 19, Number 25 Prophylaxis with oral penicillin in children with sickle cell anemia. A randomized trial MH Gaston, JI Verter, G Woods, C Pegelow, J Kelleher, G Presbury, H Zarkowsky, E Vichinsky, R Iyer, JS Lobel, and et al. PROPS I Prophylactic Penicillin Study Multicenter randomized double-blind placebo-controlled trial “Prophylactic therapy with oral penicillin by four months of age decreases the morbidity and mortality associated with pneumococcal septicemia.”

19
Vaccination: Important for adult sickle cell and all splenectomized patients!!
“Catch-up” vaccination if Prevnar series not complete

20
A fourteen year old girl with sickle cell disease comes to clinic because her left side is weak. She is immediately transferred to the ICU for exchange transfusion.

21
CT scan of stoke

22
Cerebral blood vessels

23
Sickle Cell Disease: Cross Section of Internal Carotid Artery
Normal Intimal hyperplasia Then there is large vessel disease that accounts for clinical stroke in Sickle Cell Disease, and this is an example of large vessel disease in the internal carotid artery of an 18-year-old. This slide shows the tremendous hyperplasia of the intima in this carotid, and this can grow to the point of actually occluding this very large vessel. Of course, this would then result in a stroke.

24
Age at 1st stoke in sickle syndromes
Age at first CVA. Data from the 3,647 patients used to calculate incidence rates were used to determine CVA-free survival curves. The estimated age at first CVA was significantly different for SS and SC patients (P < .001; Fig 1). The chances of having a first CVA by 20 years of age, 30 years of age, and 45 years of age were estimated at 11%, 15%, and 24%, respectively, for SS patients and 2%, 4%, and 10%, respectively, for those with SC.
. The chances of having a first CVA by 20 years of age, 30 years of age, and 45 years of age were estimated at 11%, 15%, and 24%, respectively, for SS patients and 2%, 4%, and 10%, respectively, for those with SC.")
25
Stroke Prevention Most clinical strokes occur in children with increased cerebral blood vessel flow velocities Flow measured by transcranial Doppler ultrasound STOP Trial - red cell transfusions reduce the risk of stroke in children with TCD > 200 cm/sec This diagram is from an article by Dr. Adams and his colleagues in 1990 and shows the placement of the transducer for TCD flow velocity measurements. The internal carotid, the middle cerebral, and the anterior cerebral arteries can be examined using TCD ultrasonography. Dr. Adams and his colleagues have reported the utility of TCD in predicting stroke risk for children with sickle cell anemia. Early studies showed that children with sickle cell anemia have higher baseline TCD flow velocities than normal age-matched children, thought to be related, at least in part, to their low hematocrit. In 315 children without stroke, 17.5% (55/315) had TCD flow velocity in the middle cerebral artery over 170 cm/sec and 7.9% (25/315) had TCD velocity of 200 cm/sec or greater. During 64 months of follow-up 17 patients had a stroke. Twelve of the strokes occurred in patients with TCD velocity of at least 170 cm/sec and 10 in patients with TCD velocity greater than or equal to 200 cm/sec, hence the designation conditional and abnormal. Based on these data, the STOP trial demonstrated that monthly blood transfusions significantly reduce the risk of primary stroke for children with SCA whose TCD velocity is greater than or equal to 200 cm/sec.
 had TCD flow velocity in the middle cerebral artery over 170 cm/sec and 7.9% (25/315) had TCD velocity of 200 cm/sec or greater. During 64 months of follow-up 17 patients had a stroke. Twelve of the strokes occurred in patients with TCD velocity of at least 170 cm/sec and 10 in patients with TCD velocity greater than or equal to 200 cm/sec, hence the designation conditional and abnormal. Based on these data, the STOP trial demonstrated that monthly blood transfusions significantly reduce the risk of primary stroke for children with SCA whose TCD velocity is greater than or equal to 200 cm/sec.")
26
Transcranial Doppler U/S

27
Long term treatment of sickle cell disease
Hydroxyurea Hematopoietic stem cell transplantation Gene therapy

28
Hydroxyurea Hydroxyurea decreases crises in patients with severe sickle cell disease. In 299 adults with severe sickle cell anemia: Hydroxyurea provided ~50% decrease in: frequency of hospitalization incidence of pain, acute chest syndrome, and blood transfusions In good responders: hemolysis and leukocyte counts fell hemoglobin concentrations increased Hb F increased from 5 % to 9% overall Hb F increased to 18 % in top quartile of responders N Engl J Med 1995;332:

29
Stem Cell Transplantation
Stroke risk Acute chest syndrome risk Inexorable accrual of chronic end organ damage (including CHF, pulmonary hypertension, iron overload) Transplant-related mortality Infertility Secondary malignancy Graft vs. host disease Complications of sickle cell Complications of transplantation
 Transplant-related mortality. Infertility. Secondary malignancy. Graft vs. host disease. Complications. of sickle cell. Complications. of transplantation.")
30
Gene therapy

31
Ely and Rainer NEJM 1997;336:1364

Similar presentations













 presents to the Emergency Room with a 2 day history of weakness.>")




 Hb is found in RBCs its main function is to transport O2 to tissues. Structure: 2 parts : heme + globin Globin: four chains. Heme: porphyrin.>")
: David Ginsburg, 2009 License: Unless otherwise noted, this material is made available under the terms of the Creative Commons Attribution–Noncommercial–Share.>")

