Download presentation
Presentation is loading. Please wait.

1
HEMATOPOIETIC AND ANTI- ANEMIA AGENTS February 18, 2014 Thomas M. Guenthner, PhD 407D, MSB 996-7635 tmg@uic.edu

2
Learning Objectives: 1.Basic knowledge of the epidemiology, pathophysiology, and etiology of common anemias 2.Controlling factors in erythrocyte and heme biosynthesis 3.Role of dietary iron and iron supplements in the therapy of iron-deficiency (microcytic) anemia 4. Consequences of iron overdose toxicity, and its treatment with chelating agents 5.Role of vitamin B12 and folic acid deficiency in megaloblastic (macrocytic) anemia 6.Cause of “Pernicious” anemia 7.Role of hematopoietic growth factors in erythrocyte maturation 8.Proper therapeutic role of erythropoietin 9.Consequences of improper use of erythropoietin 10.Basic knowledge of common hemoglobinopathies (Thalassemias, Sickle Cell Disease) 11.Current status of drug therapy for Sickle Cell Disease
 anemia 6.Cause of Pernicious anemia 7.Role of hematopoietic growth factors in erythrocyte maturation 8.Proper therapeutic role of erythropoietin 9.Consequences of improper use of erythropoietin 10.Basic knowledge of common hemoglobinopathies (Thalassemias, Sickle Cell Disease) 11.Current status of drug therapy for Sickle Cell Disease.")
3
Anemia - (from Ancient Greek: ἀ ναιμία (anaimia), meaning lack of blood, from ἀ ν- (an-), "not" + α ἷ μα (haima), "blood") is a decrease in the number of erythrocytes or in the normal quantity of hemoglobin in the blood. It can also include decreased oxygen-binding ability of the hemoglobin molecule due to structural abnormality.

5
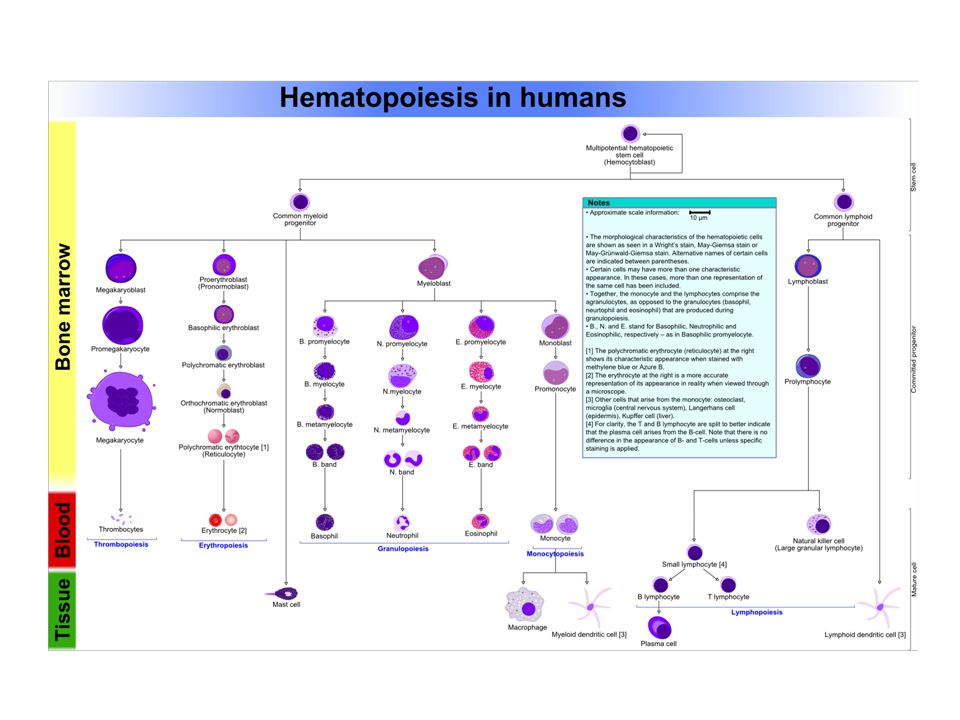
Normal total circulating erythrocyte population (adult) = 25 x 10 12 (5 x 10 6 / l) Circulating RBC’s collectively contain 2.5 - 3 g heme iron (~65% of total body iron) Life span of one RBC is ~120 days Turnover of circulating RBC’s is 1-5 x 10 6 cells/second Maturation of a circulating RBC requires ~ 7 days Erythrocyte Development
 = 25 x (5 x 10 6 / l) Circulating RBC’s collectively contain g heme iron (~65% of total body iron) Life span of one RBC is ~120 days Turnover of circulating RBC’s is 1-5 x 10 6 cells/second Maturation of a circulating RBC requires ~ 7 days Erythrocyte Development")
6
Hemoglobin Biosynthesis

7
Heme Normal Hemoglobin HbA (α 2 β 2 ) subunit subunit
 subunit subunit")
8
Pathologies associated with anemia Iron Deficiency (Microcytic) Anemia Vitamin Deficiency (Macrocytic/Megaloblastic) Anemia Anemias due to hematopoietic growth factor deficiency Functional Anemias due to hemoglobinopathies (Thalassemias, Sickle Cell Disease)
 Anemia Vitamin Deficiency (Macrocytic/Megaloblastic) Anemia Anemias due to hematopoietic growth factor deficiency Functional Anemias due to hemoglobinopathies (Thalassemias, Sickle Cell Disease)")
9
Iron Deficiency (Microcytic) Anemia RBC numbers in normal range Cells are hypochromic Mean cell size is < 80% normal Due to insufficient iron to maintain normal Hemoglobin biosynthesis
 Anemia RBC numbers in normal range Cells are hypochromic Mean cell size is < 80% normal Due to insufficient iron to maintain normal Hemoglobin biosynthesis")
10
Microcytic Anemia

11
Incidence of Microcytic Anemia

12
Iron metabolism in the Body

13
Dietary Iron requirements in different populations

14
Iron demands of pregnancy

15
Treatment of Iron Deficiency Anemia Increase Dietary Iron Oral Iron Supplement

16
Complications Hindered ability to absorb iron

17
Parenteral Iron Preparations Iron dextran Sodium ferric gluconate complex Iron sucrose

18
Complication – Iron overdose toxicity Acute Iron Overdose –Corrosion of GI epithelium –Severe acidosis –Coma –Death Chronic Iron Overdose (Hemochromatosis) –Iron deposition in tissues as hemosiderin (hemosiderosis)
 –Iron deposition in tissues as hemosiderin (hemosiderosis)")
19
Hemosiderosis

20
Antidote – Chelation Therapy Deferoxamine (Desferal) Deferasirox
 Deferasirox")
21
Macrocytic/Megaloblastic Anemia

23
Fewer, larger RBC’s Hematocrit may approximate normal value Cells are normochromic, but fewer cells = less oxygen-carrying capacity Functional Anemia Many ovoid (football-shaped) erythrocytes, up to 2 -3 x normal size
 erythrocytes, up to 2 -3 x normal size")
24
Treatment – Folate and Vitamin B12 Supplementation

25
Pernicious Anemia – Inability to absorb dietary Vitamin B12 Treatment – Parenteral B12 (Hydroxocobalamin, Cyanocobolamin)
")
26
Erythropoietin

28
Erythropoietin Supplementation

29
Erythropoietin

30
Other Hemopoietic Growth Factors Multilineage Growth Factors –Granulocyte-Myocyte Colony-Stimulating Factor (GM-CSF) Sargramostim Leukocyte Growth Factors –Granulocyte Colony-Stimulating Factor (G-CSF) Filgrastim Thrombopoietic Growth Factors –Thrombopoietin –Interleukin 11 Oprelvekin
 Sargramostim Leukocyte Growth Factors –Granulocyte Colony-Stimulating Factor (G-CSF) Filgrastim Thrombopoietic Growth Factors –Thrombopoietin –Interleukin 11 Oprelvekin")
32
Hemoglobinopathies- Sickle Cell Disease and Thalassemias Normal Hemoglobin A (α 2 β 2 ) Heme subunit subunit
 Heme subunit subunit")
33
Types of Thalassemias α Thalassemias result in decreased alpha-globin production, therefore fewer alpha- globin chains are produced, resulting in an excess of β chains in adults and excess γ chains in newborns. The excess β chains form unstable Hb tetramers, which have abnormal oxygen dissociation curves. However, even in the homozygous state this disorder will result only in a mild microcytic anemia. β o or β Thalassemia major prevents any formation of β chains, the most severe form of β thalassemia. β + or β Thalassemia intermedia allow some β chain formation to occur. In either case, there is a relative excess of α chains, but these do not form tetramers. Rather, they bind to the red cell membrane, producing membrane damage, and at high concentrations they form toxic cell aggregates. δ Thalassemia About 3% of adult hemoglobin is made of alpha and delta chains. Mutations can occur which affect the ability to produce delta chains. Hematologically, however, this is innocuous because only 2-3% of normal adult hemoglobin is hemoglobin A2. The individual will have normal hematological parameters (erythrocyte count, total hemoglobin, mean red cell volume)
.")
34
Treatment of Thalassemias No drug treatment known. Palliative treatment involves blood transfusions. Curative treatment involves bone marrow transplant. Frequent transfusion can produce iron overload toxicity.

35
Sickle Cell Disease A hemoglobinopathy involving a point mutation on the beta chain In sickle cell hemoglobin (HbS) glutamic acid in position 6 (in the beta chain) is mutated to valine. This change allows the deoxygenated form of the hemoglobin to adhere to itself and form large Hb aggregates.

36
Sickle Cell Erythrocytes

37
Treatment of Sickle Cell Disease Transfusion Bone Marrow Transplant Drugs –Hydroxyurea Increases globin gene expression and HbF production Mechanism of action not well understood Well tolerated Effective in ~60% of patients –(5-Azacytidine) Increases globin gene expression and HbF production Mechanism of action related to DNA demethylation Poorly tolerated Major cytotoxicity and carcinogenic potential Not considered useful
 Increases globin gene expression and HbF production Mechanism of action related to DNA demethylation Poorly tolerated Major cytotoxicity and carcinogenic potential Not considered useful")
38
Drug Therapy of Sickle Cell Disease Strategy involves promoting production of Fetal Hemoglobin (HbF) subunit Even 20% replacement of chain with chain provides significant relief from sickling.
 subunit Even 20% replacement of chain with chain provides significant relief from sickling.")
39
Treatment of Sickle Cell Disease Transfusion Bone Marrow Transplant Drugs –Hydroxyurea Increases globin gene expression and HbF production Mechanism of action not well understood Well tolerated Effective in ~60% of patients –(5-Azacytidine) Increases globin gene expression and HbF production Mechanism of action related to DNA demethylation Poorly tolerated Major cytotoxicity and carcinogenic potential Not considered useful
 Increases globin gene expression and HbF production Mechanism of action related to DNA demethylation Poorly tolerated Major cytotoxicity and carcinogenic potential Not considered useful")
40
Drugs to know IRON SUPPLEMENTS Ferrous Sulfate Ferrous Gluconate Iron Dextran Iron Sucrose (Deferoxamine) VITAMINS Folic Acid Vitamin B12 (Cyanocobalamin) Hydroxocobalamin HEMATOPOIETIC GROWTH FACTORS Erythropoietins Epoetin Alfa Darbepoetin Myeloid Growth Factors Filgrastim Sargramostim Thrombopoietic Growth Factors Interleukin-11 Thrombopoietin SICKLE CELL DRUGS Hydroxyurea (5-Azacytidine)
 VITAMINS Folic Acid Vitamin B12 (Cyanocobalamin) Hydroxocobalamin HEMATOPOIETIC GROWTH FACTORS Erythropoietins Epoetin Alfa Darbepoetin Myeloid Growth Factors Filgrastim Sargramostim Thrombopoietic Growth Factors Interleukin-11 Thrombopoietin SICKLE CELL DRUGS Hydroxyurea (5-Azacytidine)")
Similar presentations



 Korean J Hematol 46: 41-44 Microcytic.>")










 Hb is found in RBCs its main function is to transport O2 to tissues. Structure: 2 parts : heme + globin Globin: four globin chains (2 α.>")

 is composed of four protein chains, two α and two β globin chains arranged into.>")


 Hb is found in RBCs its main function is to transport O2 to tissues. Structure: 2 parts : heme + globin Globin: four chains. Heme: porphyrin.>")


