Download presentation
Presentation is loading. Please wait.

1
ANEMIA IN PEDIATRICS

2
Anaemia is defined as a low Hb concentration in blood, or less often, as a low haematocrit, the percentage of blood volume that consists of red blood cells. The Hb and Ht range for assessing iron deficiency are: Hb (g/dL) Ht (%) Children 6 months - 5 years Children years , Children years ,
 Ht (%) Children 6 months - 5 years Children years 11,5 34. Children years 12,0 36.")
3
Normal levels of RBCs at birth range from 5. 1 to 5
Normal levels of RBCs at birth range from 5.1 to 5.3 million/mm3 for term newborns and 4.6 to 5.3 million/mm3 for premature neonates. Because of active in utero erythropoiesis, the reticulocyte count at birth is 3 to 7% in full-term babies and 8 to 10% in premature babies. This declines to 0 to 1% by the first week of age, reflecting diminished erythropoiesis. The life span of adult erythrocytes is 120 days. RBCs in term neonate will survive between 60 and 90 days. Erythrocytes from premature neonates have considerably shorter life spans, ranging from 35 to 50 days

4
Mean Cell Volume. Early embryonic RBCs are large; diameters range from 20 to 25 µm with a mean cell volume (MCV) of 180 femtoliters (fl) or µm3. Cell size decreases gradually during development reaching 130 fl at midgestation and 115 fl at term. MCV at 1 year of age is 82 fl. The mean corpuscular hemoglobin concentration (MCHC) is fairly constant from birth through adulthood.It averages 34 pg in full-term cord blood, 35 pg on the first day of life, and 33 picograms (pg) at 1 week of age. Premature neonates, however, have higher MCHCs; values range from 40 pg at 28 weeks to 38 pg at 34 weeks
 is fairly constant from birth through adulthood.It averages 34 pg in full-term cord blood, 35 pg on the first day of life, and 33 picograms (pg) at 1 week of age. Premature neonates, however, have higher MCHCs; values range from 40 pg at 28 weeks to 38 pg at 34 weeks.")
5
RBC and Hemoglobin Physiology
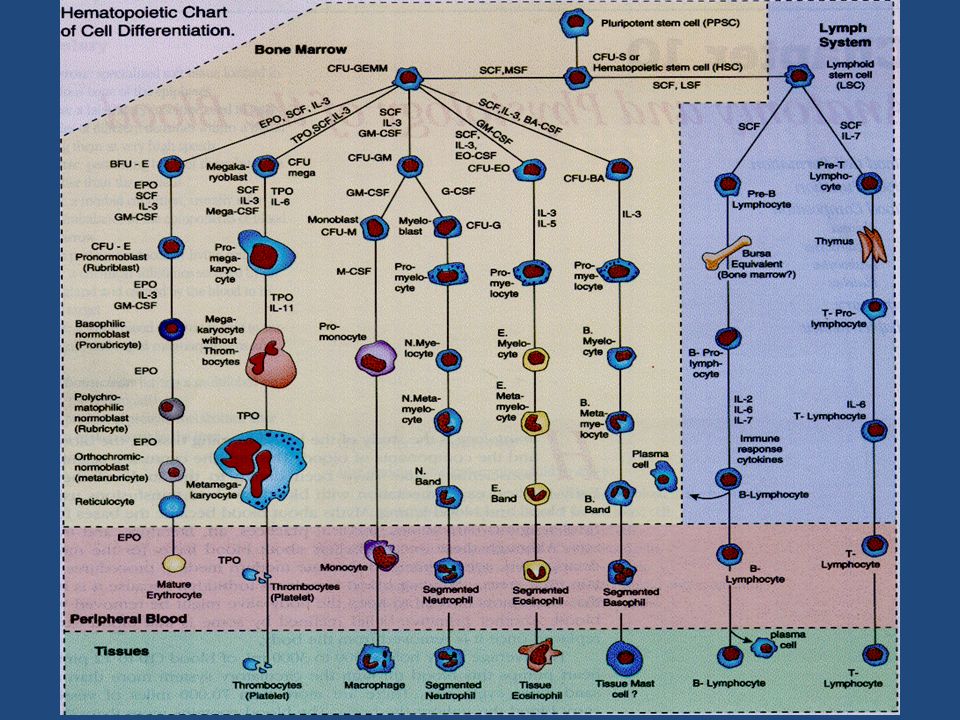
RBCs, erythrocytes, play role in the support of tissue metabolism. They contain hemoglobin, which transports oxygen to and removes carbon dioxide from tissues. RBC production involves a series of maturational steps, beginning with a pluripotent cell that differentiates into erythroid As the cells undergo maturational changes, they lose their nuclei and acquire hemoglobin. Once RBCs have achieved their normal life span, usually about 120 days, they become sequestered and destroyed in the spleen. Liberated iron is then recycled for use by the marrow in further RBC production

7
. Hemoglobin is a molecule composed of two globulin chains and four heme groups. It has been described as the respiratory protein of the RBC, related to its important role in the transport of oxygen and carbon dioxide. Hemoglobin is able to bind reversibly with oxygen, which allows it to be released to the tissues when needed. Carbon dioxide is then picked up by unbound hemoglobin for transport to the lungs and excretion. The fetus is able to produce a unique type of hemoglobin, fetal hemoglobin (HgF), which more efficiently binds and releases oxygen within the relatively hypoxic intrauterine environment.
, which more efficiently binds and releases oxygen within the relatively hypoxic intrauterine environment.")
8
Anemia refers to RBC mass, amount of hemoglobin, and/or volume of packed RBCs less than normal.
Clinically, this is determined either as a hematocrit (% of RBCs per spun whole blood sample) or hemoglobin (directly measured concentration) greater than 2 standard deviations below the normal mean for age. For children between 6 months and 2 years of age, this represents a hemoglobin <11 grams/dl or hematocrit < 33%. Hemoglobin is considered a more sensitive indicator of anemia than hematocrit, as it is not affected by variations in RBC size within the specimen; however, both are commonly utilized in clinical practice.
 or hemoglobin (directly measured concentration) greater than 2 standard deviations below the normal mean for age. For children between 6 months and 2 years of age, this represents a hemoglobin <11 grams/dl or hematocrit < 33%. Hemoglobin is considered a more sensitive indicator of anemia than hematocrit, as it is not affected by variations in RBC size within the specimen; however, both are commonly utilized in clinical practice.")
9
WHO data show that 40% of the world's population suffer from anaemia
WHO data show that 40% of the world's population suffer from anaemia. The World Health Organization calls iron deficiency the most common anemia (Centers for Disease Control) as it is estimated to affect approximately 2 billion people worldwide. The groups with the highest prevalence in pediatric age are: infants and children of 0-2 years, 48%; school children, 40%; adolescents, 30-55%; and preschool children, 25%. Anaemia occurs at a late stage of iron deficiency, after stores are depleted. The prevalence of iron deficiency, which is usually detected by low serum ferritin concentrations, is estimated to be from 2.0 to 2.5 times the prevalence of anaemia.
 as it is estimated to affect approximately 2 billion people worldwide. The groups with the highest prevalence in pediatric age are: infants and children of 0-2 years, 48%; school children, 40%; adolescents, 30-55%; and preschool children, 25%. Anaemia occurs at a late stage of iron deficiency, after stores are depleted. The prevalence of iron deficiency, which is usually detected by low serum ferritin concentrations, is estimated to be from 2.0 to 2.5 times the prevalence of anaemia.")
10
Sources of iron The sources of iron are: Birth - 6 months Breast milk alone Iron-fortified formula from birth 6 months to 1 year Infant formulas based on cow's milk contain 1.0 to 1.5 mg of iron per litre; soy-based formula and iron-fortified formula based on cow's milk contain 12 to 13 mg of iron per litre. The availability of iron from soy-based formulas appears to be lower than that from milk-based products. Iron-fortified formula ( supplementing with formula or if no breastfeeding) The iron source of fortified formulas is ferrous sulfate, which is significantly more available than the iron used in infant cereals. Iron-fortified infant cereals Iron-enriched breakfast cereals and breads Meats (poultry ) , yolk egg Fish
 The iron source of fortified formulas is ferrous sulfate, which is significantly more available than the iron used in infant cereals. Iron-fortified infant cereals. Iron-enriched breakfast cereals and breads. Meats (poultry ) , yolk egg. Fish.")
11
One litre of human milk contains only 0. 3 to 0. 5 mg of iron
One litre of human milk contains only 0.3 to 0.5 mg of iron. About 50% of the iron is absorbed, in contrast to a much smaller proportion from other foods. Term infants who are breast-fed exclusively for the first 6 months may not be at risk for iron depletion or for the development of iron deficiency. However, if solid foods are given they may compromise the bioavailability of iron from human milk. Although some term infants who are exclusively breast-fed may remain iron-sufficient until 9 months of age, a source of dietary iron is recommended starting at 6 months (or earlier if solid foods are introduced into the diet) to reduce the risk of iron deficiency.
 to reduce the risk of iron deficiency.")
12
Etiology Low iron diet Inadequate absorption of dietary iron
Growth spurt Blood loss

13
At high risk for iron deficiency are preterm infants and infants from a low socioeconomic background, low birth weight, perinatal bleeding, a low hemoglobin concentration at birth, chronic hypoxia, frequent infections, early intake of cow's milk or solid food, or both, excessive tea intake, low vitamin C or meat intake, breast-feeding for more than 6 months without supplemental iron, intake of infant formula not fortified with iron for more than 4 months without other foods.

14
Premature infants have a lower level of body iron at birth, approximately 64 mg in infants weighting 1 kg The rapid rate of postnatal growth lead to a higher requirement for dietary iron than in term infants of 2.0 to 2.5 mg/kg daily to prevent late anemia. 10% of the iron in a mixed diet is absorbed the recommended iron intake is approximately 7 mg/d for term infants aged 5 to 12 months, 6 mg/d for toddlers aged 1 to 3 years and 8 mg/d for children aged 4 to 12 years.

15
There are some factors that interfere absorbtion of iron:
High Gastric pH: hemigastrectomy, vagotomy, pernicious anemia , histamine H2 receptor blockers, calcium-based antacids Disruption of Intestinal Structure: hemigastrectomy, segments of bowel which are sometimes removed surgically, disrupting iron absorption, volvulus or intusseception, vagotomy, pernicious anemia, Inhibitors: phylates, tannins, soil clay, laundry starch, iron overload, cobalt, lead, strontium Some factors are facilitating iron absorbtion: bioavailability (heme > Fe2+> Fe3+ ), ascorbate, citrate, amino acids, iron deficiency
, ascorbate, citrate, amino acids, iron deficiency.")
16
As regarding growth spurt iron need for infants is1
As regarding growth spurt iron need for infants is1.0 mg/1,000 kcal of dietary energy; for adolescent girls, 0.8 (half of their iron requirement is needed to replace iron losses in menstruation ); adolescent boys need 0.6 mg/1,000 kcal. Some absorbtion problems can interfere iron. A major cause of anaemia is infection with malaria or other parasites. Plasmodium falciparum is the primary cause of severe malaria in regions of the world where malaria is endemic Sprue, both of the tropical and non-tropical variety (celiac disease), can also interfere with iron absorption. Degeneration of the intestinal lining cells along with chronic inflammation causes profound malabsorption.
; adolescent boys need 0.6 mg/1,000 kcal. Some absorbtion problems can interfere iron. A major cause of anaemia is infection with malaria or other parasites. Plasmodium falciparum is the primary cause of severe malaria in regions of the world where malaria is endemic Sprue, both of the tropical and non-tropical variety (celiac disease), can also interfere with iron absorption. Degeneration of the intestinal lining cells along with chronic inflammation causes profound malabsorption.")
17
Blood loss DIGESTIVE Meckel's diverticulum (persistent omphalomesenteric duct) Colonic arteriovenous malformations Arteriovenous malformations (the Osler-Weber-Rendu syndrome.) Ulcer disease are associated with Helicobacter pylori infection Gastric hiatal hernia Erosive esophogitis Milk-induced enteropathy (whole cow's milk contains proteins that often irritate the lining of the gastrointestinal tract in infants ) Parasites :the world's leading cause of gastrointestinal blood loss is parasitic infestation. Hookworm infection is caused primarily by Necator americanus or Ancylostoma duodenal Trichuris trichiura. Growth retardation, in addition to iron deficiency, occurs with heavy infestations.
 Ulcer disease are associated with Helicobacter pylori infection. Gastric hiatal hernia. Erosive esophogitis. Milk-induced enteropathy (whole cow s milk contains proteins that often irritate the lining of the gastrointestinal tract in infants ) Parasites :the world s leading cause of gastrointestinal blood loss is parasitic infestation. Hookworm infection is caused primarily by Necator americanus or Ancylostoma duodenal Trichuris trichiura. Growth retardation, in addition to iron deficiency, occurs with heavy infestations.")
18
URINARY Berger's disease, which produces relapsing episodes of gross or microscopic hematuria occurs most commonly in older children and young adults. Diffuse mesangial proliferation or focal and segmental glomerulonephritis with mesangial deposits of IgA is the most common renal pathology related to iron deficiency. Sickle cell trait occasionally develop gross hematuria. Hemoglobinuria classically is ascribed to paroxysmal nocturnal hemoglobinuria. PULMONARY Hemoptysis, chronic pulmonary infection with bronchiectasis,idiopathic pulmonary hemosiderosis, a condition characterized by recurrent pulmonary hemorrhage along with pulmonary fibrosis and right heart strain.

19
Clinical symptoms Palor of skin and mucosa
The microcytic, hypochromic anemia impairs tissue oxygen delivery, producing weakness, loss of apetite, fatigue, palpitations, and light-headedness. Iron deficiency include poor weight gain, anorexia, blood in stools, malabsorption, irritability, decreased attention span, exercise intolerance and decreased physical activity. Regarding the psychomotor development when iron deficiency progresses to anemia, performance on developmental tests is adversely affected for up to at least 3 months despite correction of the anemia with iron therapy. Among infants with severe or chronic iron deficiency, some of these abnormalities may persist indefinitely despite adequate iron therapy. The relation between iron deficiency and behavioural development is nowadays well known.

20
Iron deficiency produces significant gastrointestinal tract abnormalities. Some patients develop angular stomatitis and glossitis with painful swelling of the tongue. The flattened, atrophic lingual papillae makes the tongue smooth and shiny. A rare complication of iron deficiency is the Plummer-Vinson syndrome with the formation of a postcrycoid oesophageal web. Long-standing, severe iron deficiency affects the cells that generate the finger nails producing koilonychia. The "spoon-shaped" changes featured in many text books are rare.

21
Pica occurs variably in patients with iron deficiency
Pica occurs variably in patients with iron deficiency. The precise pathophysiology of the syndrome is unknown. Patients consume unusually laundry starch, ice, and soil clay, have abnormalities including massive hepato-splenomegaly, poor wound-healing, and a bleeding diathesis; initially had simple iron deficiency associated with pica, including geophagia. The soil contained compounds that bound both iron and zinc. The secondary zinc deficiency caused the hepatomegaly. Both clay and starch can bind iron in the gastrointestinal tract, exacerbating the deficiency. Impaired immune function is reported in subjects who are iron deficient, and there are reports that these patients are prone to infection Splenomegaly may occur with severe, persistent, untreated iron deficiency anemia and is evident in up to 15% of affected children. A systolic murmur can be heard because of the low blood viscosity and increased circulation speed of the blood

23
Laboratory tests Iron deficiency anemia lowers the number of circulating red cells (a feature of all anemias). Hb is low, the red cells are microcytic (usually less than 80 fl in size) and hypochromic. Iron deficiency alters red cell size uneven.( anyzocytosis). Mean red cell volume or mean corpuscular volume (MCV) is reduced.The range of variation in red cell size expressed as the RDW or red cell distribution width is high. membranes of iron deficient red cells are abnormally rigid. This rigidity could contribute to poikilocytic changes, seen particularly with severe iron deficiency. These small, stiff, misshapen cells are cleared by the reticuloendothelial system, contributing to the low-grade hemolysis that often accompanies iron deficiency.reticulocyte count can be under 1% or normal ( 2%-4% ). Unexplained thrombocytosis occurs frequently with platelet counts in the range of 500,000 to 700,000 cells /fl.
 and hypochromic. Iron deficiency alters red cell size uneven.( anyzocytosis). Mean red cell volume or mean corpuscular volume (MCV) is reduced.The range of variation in red cell size expressed as the RDW or red cell distribution width is high. membranes of iron deficient red cells are abnormally rigid. This rigidity could contribute to poikilocytic changes, seen particularly with severe iron deficiency. These small, stiff, misshapen cells are cleared by the reticuloendothelial system, contributing to the low-grade hemolysis that often accompanies iron deficiency.reticulocyte count can be under 1% or normal ( 2%-4% ). Unexplained thrombocytosis occurs frequently with platelet counts in the range of 500,000 to 700,000 cells /fl.")
24
In bone marrow aspirate sideroblasts are under 10%, and iron stores are low, there is an absence of stainable iron. The quantity of the iron-carrying protein, transferrin, in the circulation increases over baseline by 50% to 100%. The quantity of iron on transferrin can fall by as much as 90%. Consequently the transferrin saturation frequently declines from its usual 30% to under 10%. Ferritin is the cellular storage protein for iron and is low. The plasma ferritin value often falls to under 10% of its baseline level with significant iron deficiency. Total iron-binding capacity (TIBC) is raised. Free erythrocyte protoporphyrin (FEP) has concentrations elevate when serum iron is insufficient for RBC production. Testing stool for the presence of hemoglobin is useful in establishing gastrointestinal bleeding as the etiology of iron deficiency anemia. To detect blood loss, the patient can be placed on a strict vegetarian diet for 3-5 days and the stool can be tested for hemoglobin using a benzidine method.
 is raised. Free erythrocyte protoporphyrin (FEP) has concentrations elevate when serum iron is insufficient for RBC production. Testing stool for the presence of hemoglobin is useful in establishing gastrointestinal bleeding as the etiology of iron deficiency anemia. To detect blood loss, the patient can be placed on a strict vegetarian diet for 3-5 days and the stool can be tested for hemoglobin using a benzidine method.")
25
Positive diagnosis The diagnosis of iron deficiency is often prompted by historical features and aided by specific clinical and laboratory data.

26
Differential diagnosis
Chronic inflammation ( ex in rheumatoid arthritis ), perturb the plasma values of iron, transferrin, and ferritin. Body iron stores revealed by serum ferritin level are elevated Lead poisoning : chronic lead poisoning may produce a mild microcytosis, persons live in old houses, lead concentration is high in blood and urines. Thalasemia: in peripherial smear anemia target cells usually are present, and anisocytosis and poikilocytosis are marked, there are intraerythrocytic crystals ,bilirubin is raised, family history and clinical aspect of the child is particular in beta thalassemia. Hemoglobin electrophoresis should also be considered to rule out the presence of inherited hemoglobinopathies. Sideroblastic anemia
, perturb the plasma values of iron, transferrin, and ferritin. Body iron stores revealed by serum ferritin level are elevated. Lead poisoning : chronic lead poisoning may produce a mild microcytosis, persons live in old houses, lead concentration is high in blood and urines. Thalasemia: in peripherial smear anemia target cells usually are present, and anisocytosis and poikilocytosis are marked, there are intraerythrocytic crystals ,bilirubin is raised, family history and clinical aspect of the child is particular in beta thalassemia. Hemoglobin electrophoresis should also be considered to rule out the presence of inherited hemoglobinopathies. Sideroblastic anemia.")
27
Treatment The profilaxy of infant’s iron deficiency begins in mother’s pregnancy. The WHO recommends that all pregnant women be supplemented with 60 mg iron daily, in a pill that also usually contains 400 mg folic acid. For low born weight infants the global recommendation is to supply them with supplemental iron drops starting at 2 months of age. For developing countries the recommendation is to provide 12.5 mg of elemental iron plus 50 mg folic acid per day from age 6 months to age 12 months in regions where the anaemia prevalence is <40%, and from age 6 to 24 months where the prevalence of anaemia is > 40%. Supplements are provided for school children. Together with iron, vitamin B6, folic acid, vitamin C and vitamin A supplements are provided.

28
Treatment with iron salts
When Hb is under the value of 7gm/dl, a transfusion with red packed cells is necessary. Whole blood is a less used alternative, because of anafilactic reactions of plasma proteins, immunological antigens of lymphocytes and risk of infections. RED PACKED CELLS Treatment with iron salts Dose of elemental iron is 6-8 mg/kg/day divided in 3-4 or once daily dose before meals. Therapy is continued until fully Hb normalization; than 6-8 weeks of additional iron therapy is necessary to complete body iron stores. Although ferrous sulphate is often recommended to treat iron deficiency, frequent problems with the drug including gastrointestinal discomfort appear. Ferrous gluconate, produces fewer problems, and is preferable as the initial treatment of iron deficiency. Ascorbic acid supplementation enhances iron absorption. Polysaccharide-iron complex, is a good replacement form of iron that differs from the iron salts. Most patients tolerate this form of iron better than the iron salts, even though the 150 mg of elemental iron per tablet is substantially greater than that provided by iron salts (50 to 70 mg per tablet) Parenteral iron is available either as iron dextran or iron saccharide (commonly ferric polymaltose). There can be side effects as anaphylaxis.
 Parenteral iron is available either as iron dextran or iron saccharide (commonly ferric polymaltose). There can be side effects as anaphylaxis.")
29
Parenteral, im or iv is indicated when oral iron is poorly tolerated or gastrointestinal iron absorption is compromised Intramuscular injection of iron-dextran can be painful, and leakage into the subcutaneous tissue produces long-standing skin discoloration hages. Can also cause fever, artralgia,dyspnea, myalgia.

30
Peak reticulocytosis occurs after about 7-10 days, Hb is restored together with red blood cell number and hematocrit, and complete correction of the anemia can take 3 to 4 weeks, The hematocrit rises sufficiently in a week or two to provide symptomatic relief for most patients, serum iron, transferrin recoveres and at the end ferritin reaches normality. Human erythropoietin was one of the first agents used to correct the anemia of end-stage renal disease this hormone has provided new insight into the kinetic relationship between iron and erythropoietin in red cell production. The shifting states of storage iron contribute to the inconsistency with which erythropoietin corrects the anemia of renal failure and premature newborn anemia.

31
Follow up The hematocrit or hemoglobin should be re-evaluated in 1 month. Approximately 6 months following therapy, the hemoglobin or hematocrit should be assessed to document success of treatment and replenishment of iron stores. Severe anemia (hemoglobin < 8 g/dl) and anemia that fails to respond to adequate iron supplementation will require more intensive investigation to confirm the diagnosis and offer appropriate management.
 and anemia that fails to respond to adequate iron supplementation will require more intensive investigation to confirm the diagnosis and offer appropriate management.")
32
MEGALOBLASTIC ANEMIA Definition
The term megaloblastic anemia (MA) is used to describe a macrocytic anemia that is associated with a characteristic change-the megaloblastic change in the erythroid precursors in the bone marrow.
 is used to describe a macrocytic anemia that is associated with a characteristic change-the megaloblastic change in the erythroid precursors in the bone marrow.")
33
Causes of megaloblastic anemia
There are many causes of megaloblastic anemia, but the most common source in children occurs from a vitamin deficiency of folic acid or vitamin B-12. Other sources of megaloblastic anemia include the following: Digestive diseases These include celiac disease, chronic infectious enteritis, and enteroenteric fistulas. Pernicious anemia is a type of megaloblastic anemia caused by an inability to absorb Vitamin B-12 due to a lack of intrinsic factor in gastric (stomach) secretions. which factor enables the absorption of Vitamin B-12. malabsorption Inherited congenital folate malabsorption, a genetic disease; condition requires early intensive treatment to prevent long term problems such as mental retardation. Infection (intestinal parasites, bacterial overgrowth) Medication-induced folic acid deficiency Certain medications, specifically ones that prevent seizures, such as phenytoin, primidone, and phenobarbital, can impair the absorption of folic acid. The deficiency can usually be treated with a dietary supplement.
 secretions. which factor enables the absorption of Vitamin B-12. malabsorption Inherited congenital folate malabsorption, a genetic disease; condition requires early intensive treatment to prevent long term problems such as mental retardation. Infection (intestinal parasites, bacterial overgrowth) Medication-induced folic acid deficiency Certain medications, specifically ones that prevent seizures, such as phenytoin, primidone, and phenobarbital, can impair the absorption of folic acid. The deficiency can usually be treated with a dietary supplement.")
34
Folic acid is a B vitamin required for the production of normal red blood cells. Folic acid is present in foods such as green vegetables, liver, and yeast. It is also produced synthetically and added to many food items Foods that are rich in folic acid include the following: orange juice lentils oranges romaine lettuce chick peas (garbanzo beans) spinach Foods that are rich in folic acid and vitamin B12 include the following: liver rice eggs barley sprouts meat wheat germ poultry soy beans milk green, leafy vegetables shellfish beans fortified cereals peanuts broccoli asparagus peas
 spinach. Foods that are rich in folic acid and vitamin B12 include the following: liver. rice. eggs. barley. sprouts. meat. wheat germ. poultry. soy beans. milk. green, leafy vegetables. shellfish. beans. fortified cereals. peanuts. broccoli. asparagus. peas.")
35
Nutritional megaloblastic anemia in children occurs commonly among under-nourished or malnourished societies of tropical and subtropical countries. The commonest age is 3-18 months with maximum number of cases being in 9-12 months(1). These children are generally exclusively breast-fed by mothers who are undernourished and have poor blood levels of folate and cobalamin Intrinsic factor is a protein the body uses to absorb vitamin B12. When gastric secretions do not have enough intrinsic factor, vitamin B12 is not adequately absorbed, resulting in pernicious anemia. Absence of intrinsic factor itself is the most common cause of Vitamin B12 deficiency. Intrinsic factor is produced by cells within the stomach

36
Clinics The onset of the disease is slow and may span decades. pallor
Developmental retardation or regression is an important finding. These children are normally apathetic, not interested in surroundings and have hypotomia. Hyperpigmentation of knukles, terminal phalanges, dorsum of hand etc is seen. Tremors are described in 5-15% cases-the infantile tremor syndrome of megaloblastic anemia, hypotonia, developmental regression and tremors. Some of the cases of MA may clinically mimic cases of acute leukemia and aplastic anemia as hepatosplenomegaly (due to extramedullary hemopoiesis) of varying severity and cutaneous and other bleeding manifestations are described in 25-30% cases.abnormal paleness or lack of color of the skin decreased appetite irritability lack of energy or tiring easily (fatigue) diarrhea difficulty walking numbness or tingling in hands and feet smooth and tender tongue weak muscles
 of varying severity and cutaneous and other bleeding manifestations are described in 25-30% cases.abnormal paleness or lack of color of the skin. decreased appetite. irritability. lack of energy or tiring easily (fatigue) diarrhea. difficulty walking. numbness or tingling in hands and feet. smooth and tender tongue. weak muscles.")
37
Laboratory tests Diagnosis of MA is suggested by presence of macrocytosis. Presence of hypersegmented neutrophils also supports the diagnosis. Some cases may have circulating red cell precursors showing megaloblastic changes. Reticulocyte count is usually decreased. MCV is found to be increased (>90 fl.) What is more striking on peripheral blood examination is presence of thrombocytopenia, leucopenia and neutropenia. Thrombocytopenia is described in 50-80% cases with many of them having precariously low platelet counts. Neutropenia has been reported in 20-50% cases. Thus many cases (upto 50% in some cases) of MA may have pancytopenia - a finding which might lead to misdiagnosis of aplastic anemia (or acute leukemia if hepatosplenomegaly is also present). In some series on pancytopenia it has been observed that MA is more frequent etiological diagnosis than aplastic anemia or leukemia. The diagnosis of MA is confirmed on bone marrow examination that shows trilineal hyperplasia with megaloblastic change typically in the erythroid precursors. Reduced serum levels of B12 and folate will make the etiological diagnosis. It has been shown that serum and urinary methylmalonic acid (MMA) are increased in B12 deficiency and not in folate deficiency states.
 What is more striking on peripheral blood examination is presence of. thrombocytopenia, leucopenia and neutropenia. Thrombocytopenia is described in 50-80% cases with many of them having precariously low platelet counts. Neutropenia has been reported in 20-50% cases. Thus many cases (upto 50% in some cases) of MA may have pancytopenia - a finding which might lead to misdiagnosis of aplastic anemia (or acute leukemia if hepatosplenomegaly is also present). In some series on pancytopenia it has been observed that MA is more frequent etiological diagnosis than aplastic anemia or leukemia. The diagnosis of MA is confirmed on bone marrow examination that shows trilineal hyperplasia with megaloblastic change typically in the erythroid precursors. Reduced serum levels of B12 and folate will make the etiological diagnosis. It has been shown that serum and urinary methylmalonic acid (MMA) are increased in B12 deficiency and not in folate deficiency states.")
38
The Schilling test is performed to detect vitamin B12 absorption
The Schilling test is performed to detect vitamin B12 absorption. In the Schilling test, vitamin B12 levels are measured in the urine after the ingestion of radioactive vitamin B12. With normal absorption, the ileum (portion of the small intestine) absorbs more vitamin B12 than the body needs and excretes the excess into the urine. With impaired absorption, however, little or no vitamin B12 is excreted into the urine.
 absorbs more vitamin B12 than the body needs and excretes the excess into the urine. With impaired absorption, however, little or no vitamin B12 is excreted into the urine.")
39
Treatment Daily dose of 1mg of folic acid is more than adequate though larger doses are safe. The dose schedule if B12 therapeutic response can be obtained with a dose of 0.2 µg/kg for 2 days, but usually a 1000 µg dose is recommended which may be continued for the first 7 days. In our experience and other studies, use of this high dose has resulted in hyperexitability and tremors in some patients. Smaller dose i.e µg by initially given daily for a week and then less frequently reticulocytosis. appear, fall in MCV by 5-10 fl over 1-2 weeks, platelet count and neutrophil count tend to improve quickly, some patients may develop thrombocytosis

40
APLASTIC ANEMIAS

41
Inherited Bone Marrow Syndromes Associated with Pancytopenia,
ETIOLOGY Inherited Bone Marrow Syndromes Associated with Pancytopenia, Fanconi's Anemia Dyskeratosis Congenita Shwachman-Diamond Syndrome Cartilage-Hair Hypoplasia Pearson's Syndrome Down Syndrome Familial Marrow Dysfunction Inherited Bone Marrow Failure Syndromes Associated with Isolated Cytopenia, Diamond-Blackfan Anemia Congenital Dyserythropoietic Anemia Severe Congenital Neutropenia Inherited Thrombocytopenia Amegakaryocytic Thrombocytopenia, Thrombocytopenia with Absent Radii,

42
ETIOLOGY Aquired: * Imune * Nonimune : radiations
drugs- Chloramphenicol, phenylbutazone, and gold benzene exposure, chemicals infectious causes such as hepatitis viruses, Ebstein-Barr virus (EBV), HIV, parvovirus, and mycobacterial infections Among the acquired cytopenias paroxysmal nocturnal hemoglobinuria (PNH) is relatively rare;however, it can pose formidable management problems. Since its first recognition as a disease, PNH has been correctly classified as a hemolyticanemia; however, the frequent co-existence of other cytopenias has hinted strongly at a more complex pathogenesis. In most patients, an autoimmune mechanism has been inferred from positive responses to nontransplant therapies and laboratory data. Cytotoxic T cell attack, with production of type I cytokines, leads to hematopoietic stem cell destruction and ultimately pancytopenia. The antigen that incites disease is unknown in aplastic anemia.
, HIV, parvovirus, and mycobacterial infections. Among the acquired cytopenias paroxysmal nocturnal hemoglobinuria (PNH) is relatively rare;however, it can pose formidable management problems. Since its first recognition as a disease, PNH has been correctly classified as a hemolyticanemia; however, the frequent co-existence of other cytopenias has hinted strongly at a more complex pathogenesis. In most patients, an autoimmune mechanism has been inferred from positive responses to nontransplant therapies and laboratory data. Cytotoxic T cell attack, with production of type I cytokines, leads to hematopoietic stem cell destruction and ultimately pancytopenia. The antigen that incites disease is unknown in aplastic anemia.")
43
Inherited bone marrow failure syndromes (IBMFSs) are genetic disorders characterized by inadequate blood cell production Bone marrow failure usually presents in childhood, with petechiae, bruising, and hemorrhages due to thrombocytopenia; pallor and fatigue from anemia; and infections due to neutropenia

44
Inherited bone marrow failure syndromes (IBMFS) are genetic disorders characterized by inadequate blood cell production. Bone marrow failure (BMF) may be manifested as an isolated cytopenia (pure red cell aplasia, neutropenia, or thrombocytopenia) or as pancytopenia and the clinical picture of aplastic anemia. Other organ systems are often affected by these genetic abnormalities and result in birth defects or clinical disease in nonhematopoietic organs. Birth defects and extrahematopoietic manifestations are often characteristic and may be noticed before the onset of BMF. BMF may be present at birth (congenital BMF) or develop later in life
 or develop later in life.")
45
Fanconi's anemia is an autosomal recessive and X-linked disorder characterized by progressive bone marrow failure, congenital abnormalities, and a predisposition for malignancies. Cells from FA patients exhibit spontaneous chromosomal instability and a characteristic hypersensitivity to DNA interstrand cross-linking agents. The incidence of FA is estimated to be approximately 3 per million with a carrier frequency of 1 in 300. The clinical manifestations of FA are heterogeneous (variable penetrance and expressivity)
")
46
Specific Types of Anomalies in Fanconi's Anemia SKIN
Generalized hyperpigmentation on the trunk, neck, and intertriginous areas; café au lait spots; hypopigmented areas BODY Short stature, delicate features, small size, underweight UPPER LIMBS Thumbs: absent or hypoplastic; supernumerary, bifid, or duplicated; rudimentary; short triphalangeal, tubular, stiff, hyperextensible Radii: absent or hypoplastic (only with abnormal thumbs); absent or weak pulse Hands: clinodactyly; hypoplastic thenar eminence; six fingers; absent first metacarpal; enlarged, abnormal fingers; short fingers, transverse crease Ulnae: dysplastic
; absent or weak pulse. Hands: clinodactyly; hypoplastic thenar eminence; six fingers; absent first metacarpal; enlarged, abnormal fingers; short fingers, transverse crease. Ulnae: dysplastic.")
47
OTHER SKELETAL ANOMALIES
GONADS Males: hypogenitalia, undescended testes, hypospadias, abnormal genitalia, absent testis, atrophic testes, azoospermia, phimosis, abnormal urethra, micropenis, delayed development Females: hypogenitalia; bicornuate uterus; abnormal genitalia; aplasia of the uterus and vagina; atresia of the uterus, vagina, and ovary OTHER SKELETAL ANOMALIES Head and face: microcephaly, hydrocephalus, micrognathia, peculiar face, bird-like face, flat head, frontal bossing, scaphocephaly, sloped forehead, choanal atresia, dental abnormalities Neck: Sprengel's deformity; short, low hairline; webbed Spine: spina bifida (thoracic, lumbar, cervical, occult sacral), scoliosis, abnormal ribs, sacrococcygeal sinus, vertebral anomalies, extra vertebrae EYES Small eyes, strabismus, epicanthal folds, hypertelorism, ptosis, cataracts, astigmatism, blindness,, nystagmus, proptosis, small iris EARS Deafness (usually conductive); abnormal shape; atresia; dysplasia; low-set, large or small; infections; abnormal middle ear; absent drum; canal stenosis
, scoliosis, abnormal ribs, sacrococcygeal sinus, vertebral anomalies, extra vertebrae. EYES. Small eyes, strabismus, epicanthal folds, hypertelorism, ptosis, cataracts, astigmatism, blindness,, nystagmus, proptosis, small iris. EARS. Deafness (usually conductive); abnormal shape; atresia; dysplasia; low-set, large or small; infections; abnormal middle ear; absent drum; canal stenosis.")
48
GASTROINTESTINAL SYSTEM
KIDNEYS Ectopic or pelvic; abnormal, horseshoe, hypoplastic, or dysplastic; absent; hydronephrosis or hydroureter; infections; duplicated; rotated; reflux; hyperplasia; no function; abnormal artery GASTROINTESTINAL SYSTEM High-arched palate, atresia (esophagus, duodenum, jejunum), imperforate anus, tracheoesophageal fistula, Meckel's diverticulum, umbilical hernia, hypoplastic uvula, abnormal biliary ducts, megacolon, abdominal diastasis, Budd-Chiari syndrome LOWER LIMBS Feet: toe syndactyly, abnormal toes, flatfeet, short toes, clubfeet, six toes, supernumerary toe Legs: congenital hip dislocation, Perthes' disease, coxa vara, abnormal femur, thigh osteoma, abnormal legs CARDIOPULMONARY SYSTEM Patent ductus arteriosus, ventricular septal defect, abnormal heart, peripheral pulmonic stenosis, aortic stenosis, coarctation, absent lung lobes, vascular malformation, aortic atheromas, atrial septal defect, tetralogy of Fallot, pseudotruncus, hypoplastic aorta, abnormal pulmonary drainage, double aortic arch, cardiac myopathy OTHER ANOMALIES Slow development, hyperreflexia, Bell's palsy, central nervous system arterial malformation, stenosis of the internal carotid artery, small pituitary gland, absent corpus callosum
, imperforate anus, tracheoesophageal fistula, Meckel s diverticulum, umbilical hernia, hypoplastic uvula, abnormal biliary ducts, megacolon, abdominal diastasis, Budd-Chiari syndrome. LOWER LIMBS. Feet: toe syndactyly, abnormal toes, flatfeet, short toes, clubfeet, six toes, supernumerary toe. Legs: congenital hip dislocation, Perthes disease, coxa vara, abnormal femur, thigh osteoma, abnormal legs. CARDIOPULMONARY SYSTEM. Patent ductus arteriosus, ventricular septal defect, abnormal heart, peripheral pulmonic stenosis, aortic stenosis, coarctation, absent lung lobes, vascular malformation, aortic atheromas, atrial septal defect, tetralogy of Fallot, pseudotruncus, hypoplastic aorta, abnormal pulmonary drainage, double aortic arch, cardiac myopathy. OTHER ANOMALIES. Slow development, hyperreflexia, Bell s palsy, central nervous system arterial malformation, stenosis of the internal carotid artery, small pituitary gland, absent corpus callosum.")
50
Hematologic Abnormalities
The first hematologic abnormalities in individuals with FA are detected at a median age of 7 years. The majority of FA patients already have pancytopenia at the time of diagnosis (53%). By the age of 40, the cumulative incidence of hematologic abnormalities is 90% to 98% In rare cases, thrombocytopenia may be present at birth and progress to pancytopenia in the neonatal period or infancy Bone marrow examination generally shows reduced cellularity
. By the age of 40, the cumulative incidence of hematologic abnormalities is 90% to 98% In rare cases, thrombocytopenia may be present at birth and progress to pancytopenia in the neonatal period or infancy. Bone marrow examination generally shows reduced cellularity.")
51
FA HAS a predisposition for cancer, including leukemia and solid tumors
The risk of solid tumors and leukemia (AML) developing increases with age
 developing increases with age.")
52
Diagnosis The most widely used diagnostic test for FA is hypersensitivity to the clastogenic (chromosome-breaking) effect of diepoxybutane (DEB) or mitomycin C (MMC). The increased spontaneous chromosomal instability of FA cells leads to distinctive chromosomal breaks, gaps, and various chromatid interchanges, which were previously used as a cellular marker for FA.
 effect of diepoxybutane (DEB) or mitomycin C (MMC). The increased spontaneous chromosomal instability of FA cells leads to distinctive chromosomal breaks, gaps, and various chromatid interchanges, which were previously used as a cellular marker for FA.")
53
Clinical Management Once the diagnosis is confirmed, the family should be referred to a clinical geneticist for counseling and careful examination of family members. Genetic testing or chromosome breakage analysis should be offered to all family members. Early diagnosis is important for correct management of hematologic complications, diagnosis and appropriate treatment of coexisting congenital abnormalities and associated endocrinopathies, and identification of affected but asymptomatic family members and unaffected family members Initially, in the absence of hematologic abnormalities or clinical and laboratory signs of mild BMF, monitoring of peripheral blood values and yearly examination of bone marrow might be sufficient (see also elsewhere

54
Androgens and hematopoietic growth factors transiently improve BMF in about 50% to 60% of FA patients. Side effects of androgen therapy include masculinization, acne, hyperactivity, growth spurt followed by premature closure of the epiphyseal growth plates, liver enzyme abnormalities, hepatic adenomas, and a risk for hepatic adenocarcinomas Frequent monitoring of liver function and liver ultra-sound scans. Hematopoietic growth factors (granulocyte colony-stimulating factor [G-CSF]or granulocyte-macrophage colony-stimulating factor [GM-CSF]may transiently improve neutrophil counts and, in rare FA patients, also hemoglobin levels and platelet numbers. The use of hematopoietic growth factors in patients with clonal cytogenetic abnormalities is controversial because of the potential risk of inducing or promoting leukemia. Red cell transfusion therapy and iron chelation (after multiple red cell transfusions) are frequently used in symptomatic FA patients. Platelet transfusions may be indicated in thrombocytopenic patients with bleeding or before surgical procedures. HSCT from an HLA-matched sibling donor is accepted as the best treatment to cure BMF in FA patients and to prevent progression to MDS and AML The prognosis for FA patients without HLA-matched siblings after HSCT with unrelated donors is less favorable. Extensive malformations, a positive recipient cytomegalovirus serology, the use of androgens before transplantation, and female donors were associated with a worse outcome
 are frequently used in symptomatic FA patients. Platelet transfusions may be indicated in thrombocytopenic patients with bleeding or before surgical procedures. HSCT from an HLA-matched sibling donor is accepted as the best treatment to cure BMF in FA patients and to prevent progression to MDS and AML. The prognosis for FA patients without HLA-matched siblings after HSCT with unrelated donors is less favorable. Extensive malformations, a positive recipient cytomegalovirus serology, the use of androgens before transplantation, and female donors were associated with a worse outcome.")
55
Treatment Supportive care for patients with symptomatic anemia includes transfusions of packed red blood cells that have been leukodepleted. Thrombocytopenia istreated with platelets units; single-donor platelets are preferred to reduce the frequency of antibody formation Aplastic anemia can be effectively treated by either stem cell transplantation or immuno-suppression. Hematopoietic stem cell transplantation HSCT (bone marrow, cord blood, or peripheral blood stem cells) may cure aplastic anemia Immunosuppressive therapy using antithymocyte globulin, cyclosporine, and danazol with or without human granulocyte colony-stimulating factor and high dose cyclophosphamide. Other types of imunosupressive therapy and stem cell stimulation.: Oxymetholone (Anadrol-50) -- Anabolic and androgenic derivative of testosterone 2-4 mg/kg/day; Nandrolone decanoate (Deca-Durabolin); Prednisone : 1-2 mg/kg 4 weeks
 may cure aplastic anemia. Immunosuppressive therapy using antithymocyte globulin, cyclosporine, and danazol with or without human granulocyte colony-stimulating factor and high dose cyclophosphamide. Other types of imunosupressive therapy and stem cell stimulation.: Oxymetholone (Anadrol-50) -- Anabolic and androgenic derivative of testosterone. 2-4 mg/kg/day; Nandrolone decanoate (Deca-Durabolin); Prednisone : 1-2 mg/kg 4 weeks.")
56
Dyskeratosis congenita (DC) rare inherited bone marrow failure (BMF) syndrome with X-linked, autosomal dominant, and autosomal recessive inheritance. Classically, BMF in DC patients is associated with the mucocutaneous triad, including abnormal pigmentation, dystrophic nails, and mucosal leukoplakia. About 85% of patients with classic DC are initially found to have cytopenia of one or more lineages, and pancytopenia develops in more than 95% of patients by 40 years of age. Complications of BMF, such as hemorrhage or opportunistic infection, represent the major cause of death in patients with DC. DC is a cancer predisposition syndrome

57
Laboratory finding PERIPHERAL BLOOD
Cytopenia of one or more lineages (80%) Initial manifestation highly variable Macrocytosis with or without anemia Thrombocytopenia Neutropenia Pancytopenia Low number of circulating progenitor cells Elevated hemoglobin F Elevated von Willebrand factor
 Initial manifestation highly variable. Macrocytosis with or without anemia. Thrombocytopenia. Neutropenia. Pancytopenia. Low number of circulating progenitor cells. Elevated hemoglobin F. Elevated von Willebrand factor.")
58
BONE MARROW EXAMINATION
Hypocellular bone marrow affecting all three lineages Increased number of mast cells Dyserythropoiesis Hypocellular myelodysplastic syndrome Myelodysplastic syndrome/acute myeloid leukemia

61
Diamond-Blackfan Anemia
Diamond-Blackfan anemia (DBA) is a pure red cell aplasia with autosomal dominant inheritance that is usually seen in early infancy. DBA is associated with a reduction or absence of erythroid precursors in bone marrow, variable congenital anomalies, and a predisposition to malignancy. DBA red cells characteristically have increased adenosine deaminase activity. DBA is a rare disease with a frequency of 2 to 7 per million live births and has no ethnic or gender predilection
 is a pure red cell aplasia with autosomal dominant inheritance that is usually seen in early infancy. DBA is associated with a reduction or absence of erythroid precursors in bone marrow, variable congenital anomalies, and a predisposition to malignancy. DBA red cells characteristically have increased adenosine deaminase activity. DBA is a rare disease with a frequency of 2 to 7 per million live births and has no ethnic or gender predilection.")
62
Head, face, palate Hypertelorism, cleft palate, high-arched palate, microcephaly, micrognathia, microtia, low-set ears, low hairline, epicanthus, ptosis, flat broad nasal bridge Upper limb Triphalangeal, duplex or bifid, hypoplastic thumb; flat thenar eminence; syndactyly; absent radial artery Renal, urogenital Absent kidney, horseshoe kidney, hypospadias Cardiopulmonary Ventricular septal defect, atrial septal defect, coarctation of the aorta, complex cardiac anomalies Congenital glaucoma, strabismus, congenital cataract

63
Anemia, macrocytosis, reticulocytopenia, and absent or decreased numbers of erythroid progenitor cells in bone marrow are the major diagnostic criteria for DBA. In 13% to 20% of individuals, anemia may be present at birth Elevated levels of red cell adenosine deaminase (ADA), a critical enzyme in the purine salvage pathway, are characteristic of DBA erythropoiesis and may be found in 80% to 89% of patients
, a critical enzyme in the purine salvage pathway, are characteristic of DBA erythropoiesis and may be found in 80% to 89% of patients.")
64
TREATMENT Glucocorticoid Therapy
Steroids and red cell transfusions are the main forms of therapy. Prednisone (or prednisolone) therapy is usually initiated at a dosage of 2 mg/kg/day. For glucocorticoid responders, the prednisone dosage is slowly tapered until the patient is taking an alternate-day dosage that maintains a reasonable hemoglobin level. Many patients remain on small, alternate-day doses of steroids for years. 70% - 80% of patients with DBA are initially steroid responsive, but only 60% to 70% achieve transfusion independence Iron chelation should begin as soon as patients have increased iron stores (ferritin >1500 mg/dL Subcutaneous infusion of deferoxamine is the standard chelation therapy in patients with DBA. The recently developed oral iron chelators deferiprone, a bidentate and deferasirox, a tridentate may eventually supplant deferoxamine as the standard of iron chelation therapy
 therapy is usually initiated at a dosage of 2 mg/kg/day. For glucocorticoid responders, the prednisone dosage is slowly tapered until the patient is taking an alternate-day dosage that maintains a reasonable hemoglobin level. Many patients remain on small, alternate-day doses of steroids for years. 70% - 80% of patients with DBA are initially steroid responsive, but only 60% to 70% achieve transfusion independence. Iron chelation should begin as soon as patients have increased iron stores (ferritin >1500 mg/dL. Subcutaneous infusion of deferoxamine is the standard chelation therapy in patients with DBA. The recently developed oral iron chelators deferiprone, a bidentate and deferasirox, a tridentate may eventually supplant deferoxamine as the standard of iron chelation therapy.")
65
HEMOLYTIC ANEMIAS Hemolytic anemias = reduced red-cell life span

66
Classification of Hemolytic anemias I
Classification of Hemolytic anemias I. Red cell abnormality (Intracorpuscular factors) A. Hereditary Membrane defect (spherocytosis, elliptocytosis) Metabolic defect (Glucoze-6-Phosphate-Dehydrogenaze (G6PD) deficiency, Pyruvate kinase (PK) deficiency) Hemoglobinopathies (unstable hemoglobins, thalassemias, sickle cell anemia ) B. Acquired Membrane abnormality-paroxysmal nocturnal hemoglobinuria (PNH)
 A. Hereditary 1. Membrane defect (spherocytosis, elliptocytosis) 2. Metabolic defect (Glucoze-6-Phosphate-Dehydrogenaze (G6PD) deficiency, Pyruvate kinase (PK) deficiency) 3. Hemoglobinopathies (unstable hemoglobins, thalassemias, sickle cell anemia ) B. Acquired 1. Membrane abnormality-paroxysmal nocturnal hemoglobinuria (PNH)")
67
II. Extracorpuscular factors A. Immune hemolytic anemias 1
II. Extracorpuscular factors A. Immune hemolytic anemias Autoimmune hemolytic anemia caused by warm-reactive antibodies caused by cold-reactive antibodies Transfusion of incompatible blood B. Nonimmune hemolytic anemias Chemicals Bacterial infections, parasitic infections (malaria), venons Hemolysis due to physical trauma hemolytic - uremic syndrome (HUS) thrombotic thrombocytopenic purpura (TTP) prosthetic heart valves Hypersplenism
, venons 3. Hemolysis due to physical trauma - hemolytic - uremic syndrome (HUS) - thrombotic thrombocytopenic purpura (TTP) - prosthetic heart valves 4. Hypersplenism.")
68
DISORDERS OF RED CELL MEMBRANE
Hereditary Spherocytosis Definition;incidence Most common hereditary hemolytic anemia with dominant autosomal trait among people of Northern European origin. Phisiopathology This disorder is caused by a defective gene. The defect results in an abnormal red cell membrane so that the affected cells have a smaller surface area for their volume than normal red blood cells and resulting in cytoskeleton instability. The cells are less resistant to stresses and rupture easily.

69
Four abnormalities in red cell membrane proteins include :
spectrin deficiency combined spectrin and ankyrin deficiency band 3 deficiency protein 4.2 defects

70
Clinical signs There can be a marked heterogeneity of clinical features, ranging from an asymptomatic condition to fulminant hemolytic anemia. A family history of HS is present. Jaundice ; most prominent in newborns Pallor Shortness of breath Fatigue Weakness Irritability Enlarged spleen.

71
Lab tests Elevated reticulocyte count. Blood smear shows spherocytes, anisocytosis Complete blood count shows anemia. Peripheral smear: Howell-Jolly bodies may be present Osmotic fragility and incubated fragility test (hemolysis of HS cells may be complete at a solute concentration that causes little or no lysis of normal cells), Coombs' test - direct is negative. Coombs' test - indirect is negative. Elevated bilirubin ( indirect ) Elevated LDH.
, Coombs test - direct is negative. Coombs test - indirect is negative. Elevated bilirubin ( indirect ) Elevated LDH.")
73
Treatment; complications
Red blood cells are transfused to patients whenever Hb drops under 7gm/dl or clinical status is deteriorated. Aplastic crises (severe decrease in red blood cell production) caused by a viral infection, is due to imbalance distruction - RBC production Splenectomy usually is curative. Fatal sepsis caused by capsulated organisms (eg, Streptococcus pneumoniae, Haemophilus influenzae) is a complication in children who had a splenectomy. Bilirubin gallstones are found in approximately 50% of patients. Splenectomy for children with HS should be performed when the child is older than 6 years. Before having a splenectomy, anyone with HS should have the pneumococcal vaccine. Folic acid, an important cofactor for enzymes used in production of RBCs is given at a dose of 1 mg/day Prognosis The prognosis (outlook) after splenectomy is for a normal life and a normal life expectancy.
 caused by a viral infection, is due to imbalance distruction - RBC production. Splenectomy usually is curative. Fatal sepsis caused by capsulated organisms (eg, Streptococcus pneumoniae, Haemophilus influenzae) is a complication in children who had a splenectomy. Bilirubin gallstones are found in approximately 50% of patients. Splenectomy for children with HS should be performed when the child is older than 6 years. Before having a splenectomy, anyone with HS should have the pneumococcal vaccine. Folic acid, an important cofactor for enzymes used in production of RBCs is given at a dose of 1 mg/day. Prognosis. The prognosis (outlook) after splenectomy is for a normal life and a normal life expectancy.")
74
THALASSEMIAS

75
Classification Thalassemias are genetic disorders of hemoglobin (Hb) synthesis. Their clinical severity varies widely, ranging from mild to severe forms. Alpha thalassemia affects the alpha-globin genes. Beta thalassemia affects one or both of the beta-globin genes. In beta thalassemia minor (beta thalassemia trait or heterozygous carrier-type), one of the beta-globin genes is defective. In beta thalassemia major (homozygous beta thalassemia), the production of beta-globin chains is low because both beta-globin genes are mutated.
 synthesis. Their clinical severity varies widely, ranging from mild to severe forms. Alpha thalassemia affects the alpha-globin genes. Beta thalassemia affects one or both of the beta-globin genes. In beta thalassemia minor (beta thalassemia trait or heterozygous carrier-type), one of the beta-globin genes is defective. In beta thalassemia major (homozygous beta thalassemia), the production of beta-globin chains is low because both beta-globin genes are mutated.")
76
Pathophisiology The severe imbalance of globin chain synthesis results in ineffective erythropoiesis and severe microcytic hypochromic anemia .The excess unpaired alpha-globin chains aggregate and form precipitates that damage red cell membranes, resulting in intravascular hemolysis. Premature destruction of erythroid precursors leads to intramedullary death and ineffective erythropoiesis. The profound anemia typically is associated with erythroid hyperplasia and extramedullary hematopoiesis. Gamma chain are increased, resulting in an elevated level of Hb F. The symptoms of thalassemia intermedia reflect ineffective erythropoiesis, which leads to anemia, medullary expansion, and extramedullary hematopoiesis. Iron overload is a potential complication of thalassemia. Frequency This condition appears to be common in the Mediterranean basin, northern Africa, the Indian subcontinent.

77
Clinics Thalassemia minor usually presents as an asymptomatic mild microcytic anemia and is detected through routine blood tests. Thalassemia major is a severe anemia that presents during the first few months after birth. Thalassemia minor (beta thalassemia trait) usually is asymptomatic, and it typically is identified during routine blood count evaluation. Thalassemia major (homozygous beta thalassemia) is detected during the first few months of life, when the patient's level of fetal Hb decreases. General pallor and pale conjunctivae and fingernails indicate anemia but are not specific for hemolytic anemias Jaundice In moderately severe cases, patients or their family members may observe slight pallor, slight yellowish discoloration of the sclerae, or enlarged abdomen.
 usually is asymptomatic, and it typically is identified during routine blood count evaluation. Thalassemia major (homozygous beta thalassemia) is detected during the first few months of life, when the patient s level of fetal Hb decreases. General pallor and pale conjunctivae and fingernails indicate anemia but are not specific for hemolytic anemias. Jaundice. In moderately severe cases, patients or their family members may observe slight pallor, slight yellowish discoloration of the sclerae, or enlarged abdomen.")
78
Abnormal facies with prominent facial bones and dental malocclusions
Abnormal facies with prominent facial bones and dental malocclusions. The skull and other bones may be deformed secondary to erythroid hyperplasia with intramedullary expansion and cortical bone thinning. Modifications of liver, gall bladder, and spleen. Hepatomegaly related to significant extramedullary hematopoiesis is observed. Patients who have received blood transfusions may have hepatomegaly or chronic hepatitis due to iron overload; transfusion-associated viral hepatitis resulting in cirrhosis or portal hypertension also may be seen. The gall bladder may contain bilirubin stones formed as a result of the patient's life-long hemolytic state. Bronze skin color and diabetes result from hemochromatosis due to multiple transfusions or erroneously administered iron therapy Splenomegaly typically is observed as part of the extramedullary hematopoiesis or as a hypertrophic response related to the extravascular hemolysis. Skin ulceration

79
Iron overload cause endocrine dysfunction, especially affecting the pancreas, testes, and thyroid. The heart is a major organ that is affected by iron overload and anemia. Cardiac dysfunction in patients with thalassemia major includes conduction system defects, decreased myocardial function, and fibrosis. Some patients also develop pericarditis.

80
Differential diagnosis
Red cell membrane disorders Anemia of chronic disease Lead poisoning Sideroblastic anemia Other microcytic anemia: Complications The severe anemia resulting from this disease, if untreated, can result in high-output cardiac failure Increased iron deposition results in secondary iron overload. This overload causes clinical problems : endocrine dysfunction, liver dysfunction, cardiac dysfunction The gall bladder may contain bilirubin stones formed as a result of hemolytic state

81
Treatment The goal of long-term hypertransfusional support is to maintain the patient's Hb at 9-10 g/dL. Patients receiving transfusion therapy also require iron chelation with desferrioxamine, deferasirox. Allogeneic hematopoietic transplantation may be curative in some patients with thalassemia major. Patients with thalassemia minor rarely require splenectomy, although the development of bilirubin stones can require cholecistectomy.

82
Glucose-6-Phosphate Dehydrogenase Deficiency
The G6PD enzyme catalyzes an oxidation-reduction reaction transferring electrons from one molecule to another;the G6PD enzyme functions in catalyzing the oxidation of glucose-6-phosphate to 6-phosphogluconate, while concomitantly reducing nicotinamide adenine dinucleotide phosphate (NADP+ to NADPH).
.")
83
Incidence Highest prevalence rates are found in tropical Africa Middle East, tropical and subtropical Asia, Mediterranean areas. These include neonatal jaundice, abdominal and/or back pain, dizziness, headache, dyspnea (irregular breathing), and palpitations. Clinics Phisycal signs include neonatal jaundice, abdominal and/or back pain, dizziness, headache, dyspnea (irregular breathing), and palpitations. Neonatal jaundice is a common condition in all newborns, but when it persists, G6PD deficiency is suspected. Neonatal jaundice is a yellowish coloration of eyes, skin, and mucous membranes caused by deposition of bile salts in these tissues. This is a direct result of insufficient activity of the G6PD enzyme in the liver. In some cases, the neonatal jaundice is severe enough to cause death or permanent neurologic damage. Kernicterus is a rare complication.
, and palpitations. Clinics. Phisycal signs include neonatal jaundice, abdominal and/or back pain, dizziness, headache, dyspnea (irregular breathing), and palpitations. Neonatal jaundice is a common condition in all newborns, but when it persists, G6PD deficiency is suspected. Neonatal jaundice is a yellowish coloration of eyes, skin, and mucous membranes caused by deposition of bile salts in these tissues. This is a direct result of insufficient activity of the G6PD enzyme in the liver. In some cases, the neonatal jaundice is severe enough to cause death or permanent neurologic damage. Kernicterus is a rare complication.")
84
Hemolytic anemia is another condition which may cause problems for G6PD deficient individuals,back pain,abdominal pain, jaundice and splenomegaly may be present during a crisis. An anemic response can be induced in affected individuals by certain oxidative drugs, fava beans or infections. Most common drugs that can trigger haemolytic crisis are: acetophenetidin (phenacetin), amidopyrine (aminopyrine),aspirin, pyramidone, methylene blue,nalidixic acid, quinine, vitamin K (water soluble), cotrimoxazole, nitrofurantoin, Sulfacetamide; infections that can precipitate a hemolytic episode are viral hepatitis, pneumonia, and salmonella infections.

85
Laboratory tests Level of enzyme activity of G6PD is low CBC count ( red blood cells can present Heinz bodies) with reticulocyte count to determine the level of anemia Indirect bilirubinemia occurs Serum haptoglobin levels are decreased Abdominal ultrasound may be useful in assessing for splenomegaly and gallstones. Treatment Infants with prolonged neonatal jaundice are placed under special lights, called bili-lights, which alleviate the jaundice or require exchange transfusional procedures Identification and discontinuation of the precipitating agent is critical. Individuals are treated with oxygen and bed rest, which may afford symptomatic relief. Avoid oxidant drugs. Most patients do not need treatment.
 with reticulocyte count to determine the level of anemia. Indirect bilirubinemia occurs. Serum haptoglobin levels are decreased. Abdominal ultrasound may be useful in assessing for splenomegaly and gallstones. Treatment. Infants with prolonged neonatal jaundice are placed under special lights, called bili-lights, which alleviate the jaundice or require exchange transfusional procedures. Identification and discontinuation of the precipitating agent is critical. Individuals are treated with oxygen and bed rest, which may afford symptomatic relief. Avoid oxidant drugs. Most patients do not need treatment.")
Similar presentations