Download presentation
Presentation is loading. Please wait.

1
September 24, 2003 Microarray data analysis

2
Many of the images in this powerpoint presentation are from Bioinformatics and Functional Genomics by Jonathan Pevsner (ISBN 0-471-21004-8). Copyright © 2003 by John Wiley & Sons, Inc.John Wiley & Sons, Inc These images and materials may not be used without permission from the publisher. We welcome instructors to use these powerpoints for educational purposes, but please acknowledge the source. The book has a homepage at http://www.bioinfbook.orghttp://www.bioinfbook.org Including hyperlinks to the book chapters. Copyright notice

3
Microarray data analysis begin with a data matrix (gene expression values versus samples) Page 190
 Page 190")
4
Microarray data analysis begin with a data matrix (gene expression values versus samples) Page 190 Typically, there are many genes (>> 10,000) and few samples (~ 10)
 Page 190 Typically, there are many genes (>> 10,000) and few samples (~ 10)")
5
Microarray data analysis begin with a data matrix (gene expression values versus samples) Preprocessing Inferential statisticsDescriptive statistics Page 190
 Preprocessing Inferential statisticsDescriptive statistics Page 190")
6
Microarray data analysis: preprocessing Observed differences in gene expression could be due to transcriptional changes, or they could be caused by artifacts such as: different labeling efficiencies of Cy3, Cy5 uneven spotting of DNA onto an array surface variations in RNA purity or quantity variations in washing efficiency variations in scanning efficiency Page 191

7
Microarray data analysis: preprocessing The main goal of data preprocessing is to remove the systematic bias in the data as completely as possible, while preserving the variation in gene expression that occurs because of biologically relevant changes in transcription. A basic assumption of most normalization procedures is that the average gene expression level does not change in an experiment. Page 191

8
Data analysis: global normalization Global normalization is used to correct two or more data sets. In one common scenario, samples are labeled with Cy3 (green dye) or Cy5 (red dye) and hybridized to DNA elements on a microrarray. After washing, probes are excited with a laser and detected with a scanning confocal microscope. Page 192
 or Cy5 (red dye) and hybridized to DNA elements on a microrarray. After washing, probes are excited with a laser and detected with a scanning confocal microscope. Page 192.")
9
Data analysis: global normalization Global normalization is used to correct two or more data sets Example: total fluorescence in Cy3 channel = 4 million units Cy 5 channel = 2 million units Then the uncorrected ratio for a gene could show 2,000 units versus 1,000 units. This would artifactually appear to show 2-fold regulation. Page 192

10
Data analysis: global normalization Global normalization procedure Step 1: subtract background intensity values (use a blank region of the array) Step 2: globally normalize so that the average ratio = 1 (apply this to 1-channel or 2-channel data sets) Page 192
 Step 2: globally normalize so that the average ratio = 1 (apply this to 1-channel or 2-channel data sets) Page 192")
11
Microarray data preprocessing Some researchers use housekeeping genes for global normalization Visit the Human Gene Expression (HuGE) Index: www.HugeIndex.org Page 192
 Index: Page 192")
12
Scatter plots Useful to represent gene expression values from two microarray experiments (e.g. control, experimental) Each dot corresponds to a gene expression value Most dots fall along a line Outliers represent up-regulated or down-regulated genes Page 193
 Each dot corresponds to a gene expression value Most dots fall along a line Outliers represent up-regulated or down-regulated genes Page 193.")
13
Scatter plot analysis of microarray data Page 193

14
Brain Astrocyte Fibroblast Differential Gene Expression in Different Tissue and Cell Types

15
expression level high low up down Expression level (sample 1) Expression level (sample 2) Page 193
 Expression level (sample 2) Page 193")
16
Page 195 Log-log transformation

17
Scatter plots Typically, data are plotted on log-log coordinates Visually, this spreads out the data and offers symmetry raw ratiolog 2 ratio time behavior valuevalue t=0basal1.00.0 t=1hno change1.00.0 t=2h2-fold up2.01.0 t=3h2-fold down0.5-1.0 Page 194, 197

18
expression level high low up down Mean log intensity Log ratio Page 196

19
SNOMAD converts array data to scatter plots http://snomad.org 2-fold Log 10 (Ratio ) Mean ( Log 10 ( Intensity ) ) EXP CON EXP CON EXP > CON EXP < CON 2-fold Linear-linear plot Log-log plot Page 196-197
 Mean ( Log 10 ( Intensity ) ) EXP CON EXP CON EXP > CON EXP < CON 2-fold Linear-linear plot Log-log plot Page")
20
SNOMAD corrects local variance artifacts 2-fold Log 10 ( Ratio ) Mean ( Log 10 ( Intensity ) ) robust local regression fit residual EXP > CON EXP < CON Corrected Log 10 ( Ratio ) [residuals] Mean ( Log 10 ( Intensity ) ) Page 196-197
![SNOMAD corrects local variance artifacts 2-fold Log 10 ( Ratio ) Mean ( Log 10 ( Intensity ) ) robust local regression fit residual EXP > CON EXP < CON Corrected Log 10 ( Ratio ) [residuals] Mean ( Log 10 ( Intensity ) ) Page](http://images.slideplayer.com/16/4918193/slides/slide_20.jpg "SNOMAD corrects local variance artifacts 2-fold Log 10 ( Ratio ) Mean ( Log 10 ( Intensity ) ) robust local regression fit residual EXP > CON EXP < CON Corrected Log 10 ( Ratio ) [residuals] Mean ( Log 10 ( Intensity ) ) Page")
21
SNOMAD describes regulated genes in Z-scores Corrected Log 10 ( Ratio ) Mean ( Log 10 ( Intensity ) ) 2-fold Locally estimated standard deviation of positive ratios Z= 1 Z= -1 Locally estimated standard deviation of negative ratios Local Log 10 ( Ratio ) Z-Score Mean ( Log 10 ( Intensity ) ) Z= 5 Z= -5 Corrected Log 10 ( Ratio ) Mean ( Log 10 ( Intensity ) ) 2-fold Z= 2 Z= 1 Z= -1 Z= -2 Z= 5 Z= -5
 Mean ( Log 10 ( Intensity ) ) 2-fold Locally estimated standard deviation of positive ratios Z= 1 Z= -1 Locally estimated standard deviation of negative ratios Local Log 10 ( Ratio ) Z-Score Mean ( Log 10 ( Intensity ) ) Z= 5 Z= -5 Corrected Log 10 ( Ratio ) Mean ( Log 10 ( Intensity ) ) 2-fold Z= 2 Z= 1 Z= -1 Z= -2 Z= 5 Z= -5")
22
Inferential statistics Inferential statistics are used to make inferences about a population from a sample. Hypothesis testing is a common form of inferential statistics. A null hypothesis is stated, such as: “There is no difference in signal intensity for the gene expression measurements in normal and diseased samples.” The alternative hypothesis is that there is a difference. We use a test statistic to decide whether to accept or reject the null hypothesis. For many applications, we set the significance level to p < 0.05. Page 199

23
Inferential statistics A t-test is a commonly used test statistic to assess the difference in mean values between two groups. t = = Questions Is the sample size (n) adequate? Are the data normally distributed? Is the variance of the data known? Is the variance the same in the two groups? Is it appropriate to set the significance level to p < 0.05? Page 199 x 1 – x 2 difference between mean values variability (noise)
 adequate. Are the data normally distributed. Is the variance of the data known. Is the variance the same in the two groups. Is it appropriate to set the significance level to p < Page 199 x 1 – x 2 difference between mean values variability (noise).")
24
Inferential statistics ParadigmParametric testNonparametric Compare two unpaired groupsUnpaired t-testMann-Whitney test Compare two paired groupsPaired t-testWilcoxon test Compare 3 orANOVA more groups Page 198-200

25
Inferential statistics Is it appropriate to set the significance level to p < 0.05? If you hypothesize that a specific gene is up-regulated, you can set the probability value to 0.05. You might measure the expression of 10,000 genes and hope that any of them are up- or down-regulated. But you can expect to see 5% (500 genes) regulated at the p < 0.05 level by chance alone. To account for the thousands of repeated measurements you are making, some researchers apply a Bonferroni correction. The level for statistical significance is divided by the number of measurements, e.g. the criterion becomes: p < (0.05)/10,000 or p < 5 x 10 -6 Page 199
 regulated at the p < 0.05 level by chance alone. To account for the thousands of repeated measurements you are making, some researchers apply a Bonferroni correction. The level for statistical significance is divided by the number of measurements, e.g. the criterion becomes: p < (0.05)/10,000 or p < 5 x Page 199.")
26
Page 200 Significance analysis of microarrays (SAM) SAM-- an Excel plug-in (URL: page 202) -- modified t-test -- adjustable false discovery rate
 SAM-- an Excel plug-in (URL: page 202) -- modified t-test -- adjustable false discovery rate")
27
Page 202

28
up- regulated Page 202 down- regulated expected observed

29
Descriptive statistics Microarray data are highly dimensional: there are many thousands of measurements made from a small number of samples. Descriptive (exploratory) statistics help you to find meaningful patterns in the data. A first step is to arrange the data in a matrix. Next, use a distance metric to define the relatedness of the different data points. Two commonly used distance metrics are: -- Euclidean distance -- Pearson coefficient of correlation 203
 statistics help you to find meaningful patterns in the data. A first step is to arrange the data in a matrix. Next, use a distance metric to define the relatedness of the different data points. Two commonly used distance metrics are: -- Euclidean distance -- Pearson coefficient of correlation 203.")
30
Page 205 Data matrix (20 genes and 3 time points from Chu et al.)
")
31
Page 205 3D plot (using S-PLUS software) t=0t=0.5 t=2.0
 t=0t=0.5 t=2.0")
32
Descriptive statistics: clustering Clustering algorithms offer useful visual descriptions of microarray data. Genes may be clustered, or samples, or both. We will next describe hierarchical clustering. This may be agglomerative (building up the branches of a tree, beginning with the two most closely related objects) or divisive (building the tree by finding the most dissimilar objects first). In each case, we end up with a tree having branches and nodes. Page 204
 or divisive (building the tree by finding the most dissimilar objects first). In each case, we end up with a tree having branches and nodes. Page 204.")
33
Algorithmic Techniques Hierarchical K-Nearest Neighbors (K-Means, K-Median) Neural Networks Self-Organizing Maps Principal Component Analysis
 Neural Networks Self-Organizing Maps Principal Component Analysis")
34
Agglomerative clustering a b c d e a,b 43210 Page 206

35
a b c d e a,b d,e 43210 Agglomerative clustering Page 206

36
a b c d e a,b d,e c,d,e 43210 Agglomerative clustering Page 206

37
a b c d e a,b d,e c,d,e a,b,c,d,e 43210 Agglomerative clustering …tree is constructed Page 206

38
Divisive clustering a,b,c,d,e 43210 Page 206

39
Divisive clustering c,d,e a,b,c,d,e 43210 Page 206

40
Divisive clustering d,e c,d,e a,b,c,d,e 43210 Page 206

41
Divisive clustering a,b d,e c,d,e a,b,c,d,e 43210 Page 206

42
Divisive clustering a b c d e a,b d,e c,d,e a,b,c,d,e 43210 …tree is constructed Page 206

43
divisive agglomerative a b c d e a,b d,e c,d,e a,b,c,d,e 43210 43210 Page 206

44
Page 205

45
Page 207

46
1 1 12 Page 207 Agglomerative and divisive clustering sometimes give conflicting results, as shown here

47
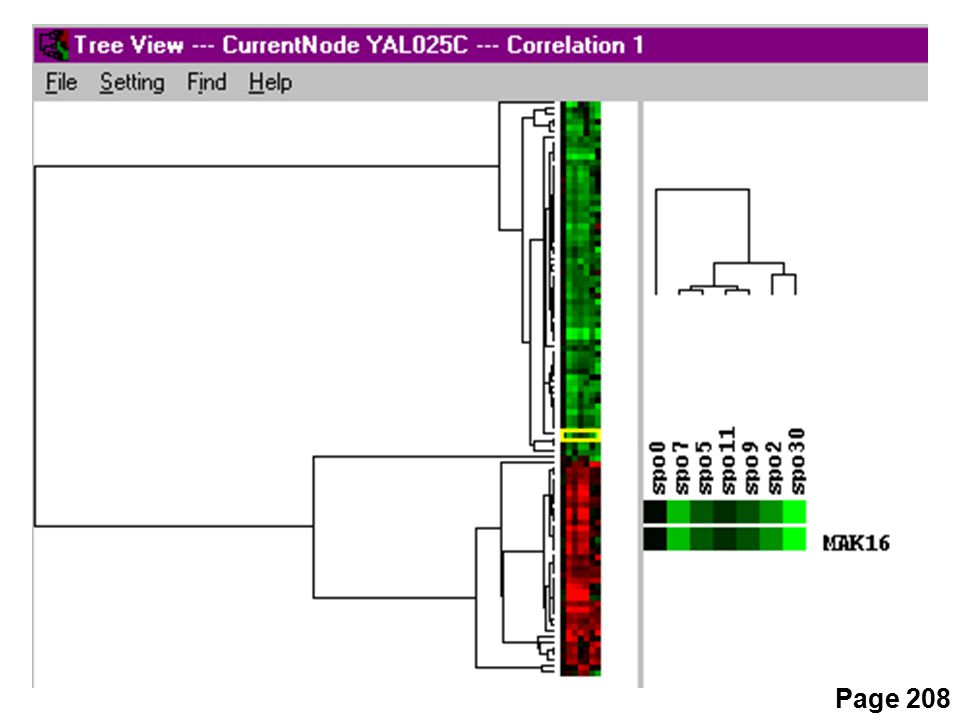
Cluster and TreeView Page 208

48
Cluster and TreeView clustering PCASOMK means Page 208

49
Cluster and TreeView Page 208

50
Cluster and TreeView Page 208

54
Page 209 Two-way clustering of genes (y-axis) and cell lines (x-axis) (Alizadeh et al., 2000)
 and cell lines (x-axis) (Alizadeh et al., 2000)")
55
Self-organizing maps (SOM) To download GeneCluster: http://www.genome.wi.mit.edu/MPR/software.html
 To download GeneCluster:")
56
Self-organizing maps (SOM) One chooses a geometry of 'nodes'-for example, a 3x2 grid http://www.genome.wi.mit.edu/MPR/SOM.html Page 210
 One chooses a geometry of nodes -for example, a 3x2 grid Page 210")
57
Self-organizing maps (SOM) The nodes are mapped into k-dimensional space, initially at random and then successively adjusted. Page 210

58
Self-organizing maps (SOM) Page 211
 Page 211")
59
Unlike k-means clustering, which is unstructured, SOMs allow one to impose partial structure on the clusters. The principle of SOMs is as follows. One chooses an initial geometry of “nodes” such as a 3 x 2 rectangular grid (indicated by solid lines in the figure connecting the nodes). Hypothetical trajectories of nodes as they migrate to fit data during successive iterations of SOM algorithm are shown. Data points are represented by black dots, six nodes of SOM by large circles, and trajectories by arrows.
. Hypothetical trajectories of nodes as they migrate to fit data during successive iterations of SOM algorithm are shown. Data points are represented by black dots, six nodes of SOM by large circles, and trajectories by arrows..")
60
Self-organizing maps (SOM) Neighboring nodes tend to define 'related' clusters. An SOM based on a rectangular grid thus is analogous to an entomologist's specimen drawer in which adjacent compartments hold similar insects.

61
Two pre-processing steps essential to apply SOMs 1. Variation Filtering: Data were passed through a variation filter to eliminate those genes showing no significant change in expression across the k samples. This step is needed to prevent nodes from being attracted to large sets of invariant genes. 2. Normalization: The expression level of each gene was normalized across experiments. This focuses attention on the 'shape' of expression patterns rather than absolute levels of expression.

62
Principal component axis #2 (10%) Principal component axis #1 (87%) PC#3: 1% C3 C4 C2 C1 N2 N3 N4 P1 P4 P2 P3 Lead (P) Sodium (N) Control (C) Legend Principal components analysis (PCA), an exploratory technique that reduces data dimensionality, distinguishes lead-exposed from control cell lines
 Principal component axis #1 (87%) PC#3: 1% C3 C4 C2 C1 N2 N3 N4 P1 P4 P2 P3 Lead (P) Sodium (N) Control (C) Legend Principal components analysis (PCA), an exploratory technique that reduces data dimensionality, distinguishes lead-exposed from control cell lines")
63
An exploratory technique used to reduce the dimensionality of the data set to 2D or 3D For a matrix of m genes x n samples, create a new covariance matrix of size n x n Thus transform some large number of variables into a smaller number of uncorrelated variables called principal components (PCs). Principal components analysis (PCA) Page 211
 Page 211.")
64
Principal components analysis (PCA): objectives to reduce dimensionality to determine the linear combination of variables to choose the most useful variables (features) to visualize multidimensional data to identify groups of objects (e.g. genes/samples) to identify outliers Page 211
 to identify outliers Page 211.")
65
Page 212 http://www.okstate.edu/artsci/botany/ordinate/PCA.htm

66
Page 212 http://www.okstate.edu/artsci/botany/ordinate/PCA.htm

67
Page 212 http://www.okstate.edu/artsci/botany/ordinate/PCA.htm

68
Page 212 http://www.okstate.edu/artsci/botany/ordinate/PCA.htm

69
Page 212

71
Chr 21 Use of PCA to demonstrate increased levels of gene expression from Down syndrome (trisomy 21) brain
 brain")
Similar presentations




 Statistical Analysis of Data Graziano and Raulin Research Methods: Chapter 5 This multimedia product and its contents.>")

 for Clustering Gene Expression Data K. Y. Yeung and W. L. Ruzzo.>")


 Dimension reduction theory (PCA/Clustering) Dimension reduction.>")






 A microarray may contain thousands of ‘spots’. Each spot contains many copies of the same DNA sequence that uniquely represents a gene from.>")


 Allows study of thousands of genes at.>")


