Download presentation
Presentation is loading. Please wait.

1
To err is human – to R is divine R from step 1 for the experimental biologist with an eye on the tomoRRow! Schraga Schwartz, Bioinformatic Workshop, June 2009

2
Outline Why R How R iRis Down syndRome Where R

3
Why ?

4
R from step 1 for the experimental biologist with an eye on the tomoRRow! R programming language is a lot like magic... except instead of spells you have functions.

5
= muggle SPSS and Excel users are like muggles. They are limited in their ability to change their environment. The way they approach a problem is constrained by how SPSS/Microsoft employed programmers thought to approach them. And they have to pay money to use these constraining softwares.

6
= wizard R users are like wizards. They can rely on functions (spells) that have been developed for them by statistical researchers, but they can also create their own. They don’t have to pay for the use of them, and once experienced enough (like Dumbledore), they are almost unlimited in their ability to change their environment.
 that have been developed for them by statistical researchers, but they can also create their own. They don’t have to pay for the use of them, and once experienced enough (like Dumbledore), they are almost unlimited in their ability to change their environment..")
7
R’s strengths Data management & manipulation Statistics Graphics Programming language Active user community Free!

8
R’s weakness Not user friendly at start. Minimal GUI. No commercial support Substantially slower than programming languages (e.g. perl, java, C++).
..")
9
R graphics: the sky's the limit! http://addictedtor.free.fr/graphiques/

10
How R?

11
R as a calculator Calculator +, -, /, *, ^, log(), exp(), sqrt(), …: (17*0.35)^(1/3) log(10) exp(1) 3^-1
, exp(), sqrt(), …: (17*0.35)^(1/3) log(10) exp(1) 3^-1")
12
Variables in R Variables are assigned using either “=“ or “ <- ” x=12.6 x [1] 12.6
![Variables in R Variables are assigned using either = or <- x=12.6 x [1] 12.6](http://images.slideplayer.com/11/3081732/slides/slide_12.jpg "Variables in R Variables are assigned using either = or <- x=12.6 x [1] 12.6")
13
Numeric vectors A vector composed of numbers. Such a vector may be created: 1. Using the c() (short for concatenate) function: y=c(3,7,9,11) > y [1] 3 7 9 11 2. Using the rep(what,how_many_times) function: y=rep(3,30) 3. Using the “:” operator, signifiying “a series of integers between” y=1:30
 (short for concatenate) function: y=c(3,7,9,11) > y [1] Using the rep(what,how_many_times) function: y=rep(3,30) 3. Using the : operator, signifiying a series of integers between y=1:30.")
14
Vector manipulation n=c(1,4,5,6,7,2,3,4,5,6) #creates a vector with the numbers in the brackets, stores it in y length(n) #number of elements n[3] #extract 3 rd element in y n[-2] #extract all of y but 2 nd element n[1:3] #extract first three element of y n[c(1,3,4)] #extract first, third, and fourth element of y n[n<4] #extract all elements in y smaller than 4 n[n<4 & n!=1] #extract element smaller than 4 AND different from 1 n[n<4 | n!=1] #extract element smaller than 4 OR different from 1
![Vector manipulation n=c(1,4,5,6,7,2,3,4,5,6) #creates a vector with the numbers in the brackets, stores it in y length(n) #number of elements n[3] #extract 3 rd element in y n[-2] #extract all of y but 2 nd element n[1:3] #extract first three element of y n[c(1,3,4)] #extract first, third, and fourth element of y n[n<4] #extract all elements in y smaller than 4 n[n<4 & n!=1] #extract element smaller than 4 AND different from 1 n[n<4 | n!=1] #extract element smaller than 4 OR different from 1](http://images.slideplayer.com/11/3081732/slides/slide_14.jpg "Vector manipulation n=c(1,4,5,6,7,2,3,4,5,6) #creates a vector with the numbers in the brackets, stores it in y length(n) #number of elements n[3] #extract 3 rd element in y n[-2] #extract all of y but 2 nd element n[1:3] #extract first three element of y n[c(1,3,4)] #extract first, third, and fourth element of y n[n<4] #extract all elements in y smaller than 4 n[n<4 & n!=1] #extract element smaller than 4 AND different from 1 n[n<4 | n!=1] #extract element smaller than 4 OR different from 1")
15
Vector manipulation… n+1 #add 1 to all elements in y n*2 #multiply by two all elements in y sum(n) mean(n) median(n) var(n) min(n) max(n) log(n) #extract logs from all variables in y sum(n[n<4]) #number of elements in y with values smaller than 4
![Vector manipulation… n+1 #add 1 to all elements in y n*2 #multiply by two all elements in y sum(n) mean(n) median(n) var(n) min(n) max(n) log(n) #extract logs from all variables in y sum(n[n<4]) #number of elements in y with values smaller than 4](http://images.slideplayer.com/11/3081732/slides/slide_15.jpg "Vector manipulation… n+1 #add 1 to all elements in y n*2 #multiply by two all elements in y sum(n) mean(n) median(n) var(n) min(n) max(n) log(n) #extract logs from all variables in y sum(n[n<4]) #number of elements in y with values smaller than 4")
16
Fuctions in R - Functions are bits of code which receive something as input (termed: arguments), and produce something as output (termed: return value). -A function can be recognized by the round brackets "()" following the function name. -The arguments of the "mean" function is a vector of numbers; the return value is their average.
 following the function name. -The arguments of the mean function is a vector of numbers; the return value is their average..")
17
Basic visualization of numbers barplot(n) plot(n) hist(n) boxplot(n) pie(n)
 plot(n) hist(n) boxplot(n) pie(n)")
18
barplot(n,col="red")
")
19
plot(n,col="red")
")
20
hist(n,col="red")
")
21
boxplot(n,col="red")
")
22
pie(n[1:3])
![pie(n[1:3])](http://images.slideplayer.com/11/3081732/slides/slide_22.jpg "pie(n[1:3])")
23
Help in R Click ? + function_name. ? barplot Help pages contain the following components: -function_name(package) – if the package is not installed, this is the time to install it and call it (using "library") -Description: brief overview -Usage -Description of arguments (input) -Details: more information -Value: value returned by the function (output) -See also: great way to learn new stuff you didn't even know you wanted to do! -Examples: Can be copy-pasted as is! Highly informative!
 – if the package is not installed, this is the time to install it and call it (using library ) -Description: brief overview -Usage -Description of arguments (input) -Details: more information -Value: value returned by the function (output) -See also: great way to learn new stuff you didn t even know you wanted to do. -Examples: Can be copy-pasted as is. Highly informative!.")
24
Other vectors Character vectors: nms=c("miriam","schragi","chaim","joc hanan","ephraim","avraham","yemima", "shakked","ayala","adi") names(n)=nms #giving names to each value in numeric vector y n["shakked"] Class Exercise: Redraw some of the previous plots with modified n!
![Other vectors Character vectors: nms=c( miriam , schragi , chaim , joc hanan , ephraim , avraham , yemima , shakked , ayala , adi ) names(n)=nms #giving names to each value in numeric vector y n[ shakked ] Class Exercise: Redraw some of the previous plots with modified n!](http://images.slideplayer.com/11/3081732/slides/slide_24.jpg "Other vectors Character vectors: nms=c( miriam , schragi , chaim , joc hanan , ephraim , avraham , yemima , shakked , ayala , adi ) names(n)=nms #giving names to each value in numeric vector y n[ shakked ] Class Exercise: Redraw some of the previous plots with modified n!")
25
The paste() function Concatenates different characters into a single character, separated by the variable defined by sep argument (default: sep=" ") paste("To","err","is human.","To R is","divine!",sep="_")
 function Concatenates different characters into a single character, separated by the variable defined by sep argument (default: sep= ) paste( To , err , is human. , To R is , divine! ,sep= _ )")
26
Boolean vectors! Boolean vectors: b=c(TRUE,FALSE,TRUE,FALSE,TRUE,TRU E)
")
27
Factor vectors (We love factors!) f=as.factor(c("stupid","stupid","s mart","stupid","imbecile","smart ","smart","imbecile")) levels(f) #possible values a variable in y can have summary(f) #provides the number of time each factor occurs Class Exercise: Compare summary(n), summary(b), and summary(f) – note difference in output!
 f=as.factor(c( stupid , stupid , s mart , stupid , imbecile , smart , smart , imbecile )) levels(f) #possible values a variable in y can have summary(f) #provides the number of time each factor occurs Class Exercise: Compare summary(n), summary(b), and summary(f) – note difference in output!")
28
The data.frame Class (We also love data.frames!) A data.frame is simply a table Each column may be of a different class (i.e. one column may be numeric, another may be a character, a third may be boolean and a fourth may be a factor) All rows in a given column must be of the same class The number of rows in each column must be identical.
 All rows in a given column must be of the same class The number of rows in each column must be identical..")
29
Iris database Petal (עלה כותרת) Sepal (עלה גביע)
 Sepal (עלה גביע)")
30
The iris dataset

31
The fascinating questions What are typical lengths and widths of sepals and petals? Do these change from one family of irises to another? Do longer petals tend to be wider? Do longer petals tend to correlate with longer (or wider) sepals? Do such correlations change from one family of irises to another?
 sepals. Do such correlations change from one family of irises to another .")
32
Playing with data frames - I 1. Set the work directory to the directory you're working in: setwd("F:/presentations/R presentation") (Note: getwd() tells you which directory you're in) 2. Load the table you want to work with (make sure you saved it as tab delimited file!): ir=read.table(file="iris_dataset.txt",sep="\t",header=T) #loads iris_dataset.txt into variable "ir". Assumes that the file is tab delimited, and that the first line is a header.
 (Note: getwd() tells you which directory you re in) 2. Load the table you want to work with (make sure you saved it as tab delimited file!): ir=read.table(file= iris_dataset.txt ,sep= \t ,header=T) #loads iris_dataset.txt into variable ir . Assumes that the file is tab delimited, and that the first line is a header..")
33
Playing with data frames II class(ir) #shows the class of ir dim(ir) #returns the number of rows and columns in ir ir[1,2] #first line, second column in ir ir[1,] #all columns in first line in ir ir[,1] #all rows in first column of ir ir$seplen #same as above ir[,"seplen"] #same as above ir[,c("seplen","sepwid")] OR ir[,1:2] #first two columns of ir summary(ir) #each of the columns is summarized according to its class
![Playing with data frames II class(ir) #shows the class of ir dim(ir) #returns the number of rows and columns in ir ir[1,2] #first line, second column in ir ir[1,] #all columns in first line in ir ir[,1] #all rows in first column of ir ir$seplen #same as above ir[, seplen ] #same as above ir[,c( seplen , sepwid )] OR ir[,1:2] #first two columns of ir summary(ir) #each of the columns is summarized according to its class](http://images.slideplayer.com/11/3081732/slides/slide_33.jpg "Playing with data frames II class(ir) #shows the class of ir dim(ir) #returns the number of rows and columns in ir ir[1,2] #first line, second column in ir ir[1,] #all columns in first line in ir ir[,1] #all rows in first column of ir ir$seplen #same as above ir[, seplen ] #same as above ir[,c( seplen , sepwid )] OR ir[,1:2] #first two columns of ir summary(ir) #each of the columns is summarized according to its class")
34
Playing with data frames - III ir$seplen>6 #returns a boolean vector with TRUE and FALSE values depending on whether seplen is greater than 6 ir[ir$seplen>6,] #returns a subset of ir containing all columns of all rows in which seplen is greater than 6 ir[ir$seplen>6,c("seplen","sepwid")] #returns same rows as above, but only "seplen" and "sepwid" columns ir[ir$seplen>6 & ir$sepwid >3,c("seplen","sepwid")] #returns same columns as above, but only rows in which seplen is greater than 6 and sepwid is greater than 3
![Playing with data frames - III ir$seplen>6 #returns a boolean vector with TRUE and FALSE values depending on whether seplen is greater than 6 ir[ir$seplen>6,] #returns a subset of ir containing all columns of all rows in which seplen is greater than 6 ir[ir$seplen>6,c( seplen , sepwid )] #returns same rows as above, but only seplen and sepwid columns ir[ir$seplen>6 & ir$sepwid >3,c( seplen , sepwid )] #returns same columns as above, but only rows in which seplen is greater than 6 and sepwid is greater than 3](http://images.slideplayer.com/11/3081732/slides/slide_34.jpg "Playing with data frames - III ir$seplen>6 #returns a boolean vector with TRUE and FALSE values depending on whether seplen is greater than 6 ir[ir$seplen>6,] #returns a subset of ir containing all columns of all rows in which seplen is greater than 6 ir[ir$seplen>6,c( seplen , sepwid )] #returns same rows as above, but only seplen and sepwid columns ir[ir$seplen>6 & ir$sepwid >3,c( seplen , sepwid )] #returns same columns as above, but only rows in which seplen is greater than 6 and sepwid is greater than 3")
35
Visualization hist(ir$seplen) #histogram of seplen
 #histogram of seplen")
36
Visualization - II hist(ir$seplen,30) #histogram of seplen
 #histogram of seplen")
37
Visualization - III mean_seplen=mean(ir$seplen) hist(ir$seplen,20,col="light blue", main ="Distribution of Septal lengths", xlab ="Lengths of septal (cm)", sub =paste("Mean septal length is",mean_seplen))
 hist(ir$seplen,20,col= light blue , main = Distribution of Septal lengths , xlab = Lengths of septal (cm) , sub =paste( Mean septal length is ,mean_seplen))")
38
The tapply() function Suppose you want to obtain average ages of patients (a numeric) variable, as a function of their gender (a factor) variable. And suppose the data is stored in the data frame data. The magic spell is: tapply(data$age,data$gender,mean) The tapply function – receives three parameters: -A numeric distribution -A factor variable, dividing the numeric distribution into groups -A function (mean,min,max,sd,sum)
 The tapply function – receives three parameters: -A numeric distribution -A factor variable, dividing the numeric distribution into groups -A function (mean,min,max,sd,sum).")
39
mean_per_species=tapply(ir$seplen,ir$species,mean) #calculates the mean value of ir$seplen after dividing it into three groups based on ir$species barplot(mean_per_species,col="red") Visualization - IV
 #calculates the mean value of ir$seplen after dividing it into three groups based on ir$species barplot(mean_per_species,col= red ) Visualization - IV")
40
Adding packages

41
Select mirror

42
Select library

43
Class exercise Install the following three libraries: gplots, lattice,car These libraries will be used in subsequent examples.

44
Visualization - V sd_per_species=tapply(ir$seplen,ir$species,sd) #caculate standard deviation library(gplots) #loads all functions in gplots into workspace (including the barplot2 function) barplot2(mean_per_species, plot.ci = T, ci.l = mean_per_species-sd_per_species, ci.u = mean_per_species+sd_per_species,col="red",ylab="Mean septal lengths")
 #caculate standard deviation library(gplots) #loads all functions in gplots into workspace (including the barplot2 function) barplot2(mean_per_species, plot.ci = T, ci.l = mean_per_species-sd_per_species, ci.u = mean_per_species+sd_per_species,col= red ,ylab= Mean septal lengths )")
45
Visualization - VI library(gplots) plotmeans(ir$seplen~ir$species,xlab="species",ylab=" Sepal length")
 plotmeans(ir$seplen~ir$species,xlab= species ,ylab= Sepal length )")
46
Looking at correlations plot(ir$petlen,ir$petwid) #plotting one set of numbers as a function of another
 #plotting one set of numbers as a function of another")
47
Arguments of the plot function Some parameters of plot() function (get more by typing "? plot.default"): x – x values (defaults 1:number of points) y – the distribution type – type: can be either "l" (line), "p" (points) or more pch – type of bullets (values from 19-25) col – color (either numbers of names of colors) – can receive multiple colors lwd – line width lty – line type xlab,ylab – X and Y labels main, sub – main title (top of chart) and subtitle (beneath the X label)
: x – x values (defaults 1:number of points) y – the distribution type – type: can be either l (line), p (points) or more pch – type of bullets (values from 19-25) col – color (either numbers of names of colors) – can receive multiple colors lwd – line width lty – line type xlab,ylab – X and Y labels main, sub – main title (top of chart) and subtitle (beneath the X label).")
48
More sophisticated plotting plot(ir$petlen,ir$petwid,col=as.numeric(ir$species),p ch=19,xlab="Petal width",ylab="Petal length")
,p ch=19,xlab= Petal width ,ylab= Petal length )")
49
And more sophisticated plot, with legend and P values stat=cor.test(ir$petlen,ir$petwid) rval=stat$estimate pval=stat$p.value plot(ir$petlen,ir$petwid,col=as.numeric(ir$species),pch=19,xlab=" Petal width",ylab="Petal length",main=paste("R=",rval," ; P=",pval,sep="")) legend(x="topleft",legend=levels(ir$species),col=1:3,lty=1,lwd=2) #adding a legend
 rval=stat$estimate pval=stat$p.value plot(ir$petlen,ir$petwid,col=as.numeric(ir$species),pch=19,xlab= Petal width ,ylab= Petal length ,main=paste( R= ,rval, ; P= ,pval,sep= )) legend(x= topleft ,legend=levels(ir$species),col=1:3,lty=1,lwd=2) #adding a legend")
50
Plotting correlations as a function of a third factor variable library("lattice") xyplot(ir$seplen ~ ir$sepwid | ir$species)
 xyplot(ir$seplen ~ ir$sepwid | ir$species)")
51
Looking at everything as a function of everything else pairs(iris[,1:4]) pairs(iris[,1:4],col=iris$Species,upper.panel=NULL)
![Looking at everything as a function of everything else pairs(iris[,1:4]) pairs(iris[,1:4],col=iris$Species,upper.panel=NULL)](http://images.slideplayer.com/11/3081732/slides/slide_51.jpg "Looking at everything as a function of everything else pairs(iris[,1:4]) pairs(iris[,1:4],col=iris$Species,upper.panel=NULL)")
52
Even more sophisticated… library(car) scatterplot.matrix(iris[,1:4],groups=iris$Species,ellipse= T,levels=0.95,upper.panel=NULL, smooth=F)
![Even more sophisticated… library(car) scatterplot.matrix(iris[,1:4],groups=iris$Species,ellipse= T,levels=0.95,upper.panel=NULL, smooth=F)](http://images.slideplayer.com/11/3081732/slides/slide_52.jpg "Even more sophisticated… library(car) scatterplot.matrix(iris[,1:4],groups=iris$Species,ellipse= T,levels=0.95,upper.panel=NULL, smooth=F)")
53
And more (for the highly motivated or extremly bored…) upperpanel.cor <- function(x, y,method="pearson",digits=2,...) { points(x,y,type="n"); usr <- par("usr"); on.exit(par(usr)) par(usr = c(0, 1, 0, 1)); correl <- cor.test(x, y,method=method); r=correl$estimate; pval=correl$p.value; color="black"; if (pval<0.05) color="blue"; txt <- format(r,digits=2) pval <- format(pval,digits=2) txt <- paste("r=", txt, "\npval=",pval,sep="") text(0.5, 0.5, txt,col=color) } scatterplot.matrix(iris[,1:4],groups=iris$Species,ellipse=T,level s=0.95,upper.panel=upperpanel.cor,cex=0.3,smooth=F,main="This is cool!!!")
![And more (for the highly motivated or extremly bored…) upperpanel.cor <- function(x, y,method= pearson ,digits=2,...) { points(x,y,type= n ); usr <- par( usr ); on.exit(par(usr)) par(usr = c(0, 1, 0, 1)); correl <- cor.test(x, y,method=method); r=correl$estimate; pval=correl$p.value; color= black ; if (pval<0.05) color= blue ; txt <- format(r,digits=2) pval <- format(pval,digits=2) txt <- paste( r= , txt, \npval= ,pval,sep= ) text(0.5, 0.5, txt,col=color) } scatterplot.matrix(iris[,1:4],groups=iris$Species,ellipse=T,level s=0.95,upper.panel=upperpanel.cor,cex=0.3,smooth=F,main= This is cool!!! )](http://images.slideplayer.com/11/3081732/slides/slide_53.jpg "And more (for the highly motivated or extremly bored…) upperpanel.cor <- function(x, y,method= pearson ,digits=2,...) { points(x,y,type= n ); usr <- par( usr ); on.exit(par(usr)) par(usr = c(0, 1, 0, 1)); correl <- cor.test(x, y,method=method); r=correl$estimate; pval=correl$p.value; color= black ; if (pval<0.05) color= blue ; txt <- format(r,digits=2) pval <- format(pval,digits=2) txt <- paste( r= , txt, \npval= ,pval,sep= ) text(0.5, 0.5, txt,col=color) } scatterplot.matrix(iris[,1:4],groups=iris$Species,ellipse=T,level s=0.95,upper.panel=upperpanel.cor,cex=0.3,smooth=F,main= This is cool!!! )")
54
Final output

55
Saving Graphics to Files Before running the visualizing function, redirect all plots to a file of a certain type. Possibilities: –jpeg(filename) –png(filename) –pdf(filename) –postscript(filename) After running the visualization function, close graphic device using dev.off()
 –png(filename) –pdf(filename) –postscript(filename) After running the visualization function, close graphic device using dev.off().")
56
Saving graphics Example: pdf("F:/test.pdf") barplot(1:10,col="red") dev.off() Note:Different graphic functions can also receive arguments regarding width and height of canvas. Use "?" + function name (e.g. ?jpeg to obtain arguments)
.")
57
Statistics t.test #Student t test wilcox.test #Mann-Whitney test kruskal.test #Kruskal-Wallis rank sum test chisq.test #chi squared test cor.test #pearson/spearman correlations lm(),glm() #linear and generalized linear models p.adjust #adjustment of P values to multiple testing using FDR, bonferroni, or whatnot…
,glm() #linear and generalized linear models p.adjust #adjustment of P values to multiple testing using FDR, bonferroni, or whatnot…")
58
Down Syndrome

59
The fascinating research question Do genes from any particular chromosome alter their expression levels in Down syndrome?

60
GEO database: A paradise of numbers

61
Getting the data!

62
Loading the data (look at it first!) setwd("F:/Presentations/R presentation/") #sets the work directory a=read.table(file="GSE5390_series_matrix.txt ",sep="\t",header=T,comment.char="!") #loads the gene expression values and stores them in a names(a)=c("id","down1","down2","down3","dow n4","down5","down6","down7","healty1","hea lty2","healty3","healty4","healty5","healt y6","healty7","healty8") #give informative names to columns in a
 setwd( F:/Presentations/R presentation/ ) #sets the work directory a=read.table(file= GSE5390_series_matrix.txt ,sep= \t ,header=T,comment.char= ! ) #loads the gene expression values and stores them in a names(a)=c( id , down1 , down2 , down3 , dow n4 , down5 , down6 , down7 , healty1 , hea lty2 , healty3 , healty4 , healty5 , healt y6 , healty7 , healty8 ) #give informative names to columns in a")
63
The merge() function a= b= merge(a,b,by="name") OR merge(a,b,by.x="name",by.y="name")
 function a= b= merge(a,b,by= name ) OR merge(a,b,by.x= name ,by.y= name )")
64
Merging data convert=read.table(file="convert_affyprobes_ 2_chromosome_location_from_UCSC.txt",sep=" \t",header=T) b=merge(a,convert,by="id") #merges a and convert by the columns indicated by the by arguments. In other words, the column "id" in "a" is compared to the column "id" in "convert". Only lines in which the two values are identical are retained, yielding a new data frame with shared values & shared information.

65
Assign informative names downcols=2:8 healthycols=9:16 allarraycols=c(downcols,healthycols)
")
66
Calculate Fold Change between disease and healthy Step 1: calculate mean expression values for all patients with Down syndrome b$meandown=apply(b[,downcols],1,mean) Step 2: calculate mean expression values for all healthy subjects b$meanhealthy=apply(b[,healthycols],1,mean) Step 3: Calculate difference between the two (since data is log transformed) b$dif=b$meandown-b$meanhealthy Step 4: anti-log the fold change b$foldchange=2^b$dif
![Calculate Fold Change between disease and healthy Step 1: calculate mean expression values for all patients with Down syndrome b$meandown=apply(b[,downcols],1,mean) Step 2: calculate mean expression values for all healthy subjects b$meanhealthy=apply(b[,healthycols],1,mean) Step 3: Calculate difference between the two (since data is log transformed) b$dif=b$meandown-b$meanhealthy Step 4: anti-log the fold change b$foldchange=2^b$dif](http://images.slideplayer.com/11/3081732/slides/slide_66.jpg "Calculate Fold Change between disease and healthy Step 1: calculate mean expression values for all patients with Down syndrome b$meandown=apply(b[,downcols],1,mean) Step 2: calculate mean expression values for all healthy subjects b$meanhealthy=apply(b[,healthycols],1,mean) Step 3: Calculate difference between the two (since data is log transformed) b$dif=b$meandown-b$meanhealthy Step 4: anti-log the fold change b$foldchange=2^b$dif")
67
Calculate P values Step 1: Create function which receives a line as input, and knows how to break it up into disease and control groups and yield a p value GetPval=function(line) { ttest=t.test(line[downcols-1],line[healthycols- 1]) ttest$p.value } Step 2: Apply this function to all rows of the data frame b$pval=apply(b[,allarraycols],1,GetPval) Step 3: Adjust P value to multiple testing b$adjustedPval=p.adjust(b$pval,method="fdr")
![Calculate P values Step 1: Create function which receives a line as input, and knows how to break it up into disease and control groups and yield a p value GetPval=function(line) { ttest=t.test(line[downcols-1],line[healthycols- 1]) ttest$p.value } Step 2: Apply this function to all rows of the data frame b$pval=apply(b[,allarraycols],1,GetPval) Step 3: Adjust P value to multiple testing b$adjustedPval=p.adjust(b$pval,method= fdr )](http://images.slideplayer.com/11/3081732/slides/slide_67.jpg "Calculate P values Step 1: Create function which receives a line as input, and knows how to break it up into disease and control groups and yield a p value GetPval=function(line) { ttest=t.test(line[downcols-1],line[healthycols- 1]) ttest$p.value } Step 2: Apply this function to all rows of the data frame b$pval=apply(b[,allarraycols],1,GetPval) Step 3: Adjust P value to multiple testing b$adjustedPval=p.adjust(b$pval,method= fdr )")
68
Saving data frames to a file write.table(b,file="DownWithPvals.txt",sep=" \t",row.names=F,col.names=T) #generates a tab- delimited file with column names, without row names containing the data in the data frame b
 #generates a tab- delimited file with column names, without row names containing the data in the data frame b")
69
Finding significant events sigs=b[b$foldchange>1.75 & b$adjustedPval<0.01,] #finding events with significant fold change and significant P values sigs=sigs[order(sigs$adjustedPval,decreasing=T),] #sorting table based on P values
![Finding significant events sigs=b[b$foldchange>1.75 & b$adjustedPval<0.01,] #finding events with significant fold change and significant P values sigs=sigs[order(sigs$adjustedPval,decreasing=T),] #sorting table based on P values](http://images.slideplayer.com/11/3081732/slides/slide_69.jpg "Finding significant events sigs=b[b$foldchange>1.75 & b$adjustedPval<0.01,] #finding events with significant fold change and significant P values sigs=sigs[order(sigs$adjustedPval,decreasing=T),] #sorting table based on P values")
70
Finding and plotting % significantly over/under expressed genes per chromosome percentages=summary(sigs$chr)*100/sum mary(b$chr) #divides the number of times each chrosome appears in "sigs" by number of time it appears in original data barplot(percentages,las=3,col="light blue",ylab="% significant genes",main="To R is divine!") #barplot depicting the percentage of genes from each chromosome within sig
*100/sum mary(b$chr) #divides the number of times each chrosome appears in sigs by number of time it appears in original data barplot(percentages,las=3,col= light blue ,ylab= % significant genes ,main= To R is divine! ) #barplot depicting the percentage of genes from each chromosome within sig")
72
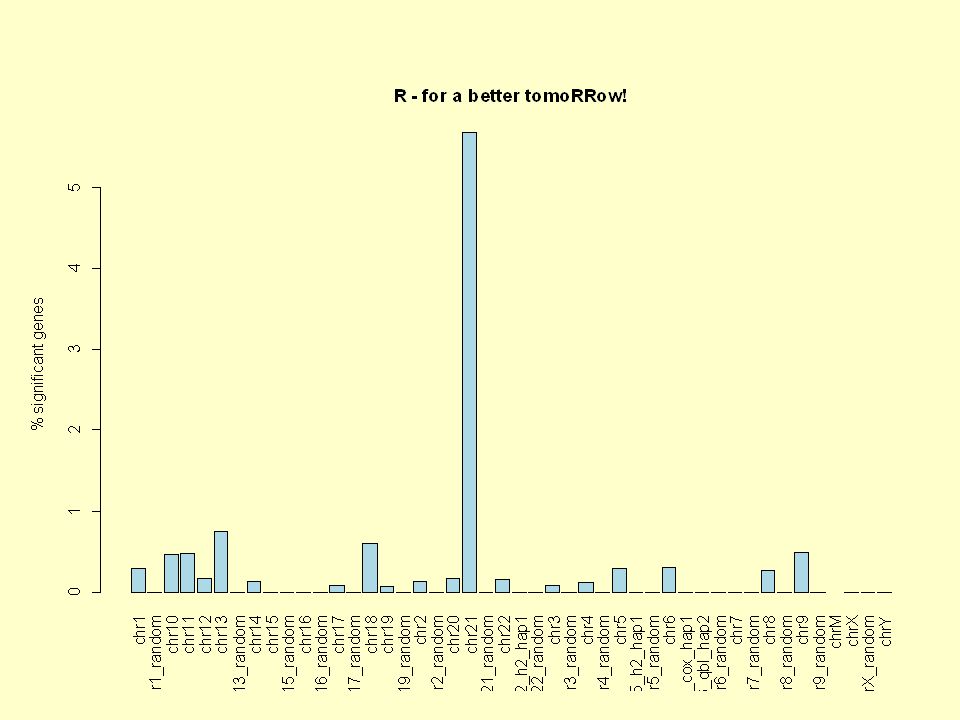
Even better plot… validchrs=c(paste("chr",1:22,sep =""),"chrX","chrY") percentages=percentages[validchr s] barplot(percentages,las=3,col="l ight blue",ylab="% significant genes",main="R - for a better tomoRRow!")
![Even better plot… validchrs=c(paste( chr ,1:22,sep = ), chrX , chrY ) percentages=percentages[validchr s] barplot(percentages,las=3,col= l ight blue ,ylab= % significant genes ,main= R - for a better tomoRRow! )](http://images.slideplayer.com/11/3081732/slides/slide_72.jpg "Even better plot… validchrs=c(paste( chr ,1:22,sep = ), chrX , chrY ) percentages=percentages[validchr s] barplot(percentages,las=3,col= l ight blue ,ylab= % significant genes ,main= R - for a better tomoRRow! )")
73
Results…

74
Volcano plots: P values as a measure of fold change plot(log2(b$foldchange),- log2(b$pval),col=(b$chr=="chr21")+1,pch=19,xlab="log fold- change",ylab="-log P value") legend(x="topleft",legend=c("non chr 21","chr 21"),lty=1,col=1:2,lwd=3) abline(h=-log2(0.001),col="blue",lty=3) abline(v=c(log2(1.75),-log2(1.75)),col="blue",lty=3) text(2,17,"Significantly\nOver-represented",col="blue") text(-1.4,17,"Significantly\nUnder-represented",col="blue") abline() function: adds either horizontal or vertical line/s (as well as more sophisticated stuff as well), depending on whether the "h" or "v" arguments are populated text() function: receives x,y coordinates |on plot, as well as text to plot
,- log2(b$pval),col=(b$chr== chr21 )+1,pch=19,xlab= log fold- change ,ylab= -log P value ) legend(x= topleft ,legend=c( non chr 21 , chr 21 ),lty=1,col=1:2,lwd=3) abline(h=-log2(0.001),col= blue ,lty=3) abline(v=c(log2(1.75),-log2(1.75)),col= blue ,lty=3) text(2,17, Significantly\nOver-represented ,col= blue ) text(-1.4,17, Significantly\nUnder-represented ,col= blue ) abline() function: adds either horizontal or vertical line/s (as well as more sophisticated stuff as well), depending on whether the h or v arguments are populated text() function: receives x,y coordinates |on plot, as well as text to plot")
75
Volcano plot

76
A particular R strength: genetics Bioconductor is a suite of additional functions and some 200 packages dedicated to analysis, visualization, and management of genetic data Much more functionality than software released by Affy or Illumina

77
Where R?

78
R homepage: http://www.r- project.org/http://www.r- project.org/

79
Choose server…

80
Click on “Windows”

81
Click “base”

82
Click on “Download” link and follow installation guidelines…

83
There you R!

84
Installing Tinn-R Go to: http://www.sciviews.org/Tinn-R/http://www.sciviews.org/Tinn-R/ Scroll to bottom of page

85
Loading R from within Tinn-R

86
Configuring Tinn-R hotkeys

87
Write text in Tinn-R; send to R

88
Final Tips Use http://www.rseek.org/ & google for finding help on what you wanthttp://www.rseek.org/ Know your objects’ classes: class(x) Know your functions arguments. Use "? function_name" to learn what arguments a function receives & what its return values are. Each help files provides examples, which can be copy-pasted into R as is. Extremely useful! MOST IMPORTANT - the more time you spend using R, the more comfortable you become with it. DESPAIR NOT – and you will never look back!

89
Final Words of Warning “Using R is a bit akin to smoking. The beginning is difficult, one may get headaches and even gag the first few times. But in the long run,it becomes pleasurable and even addictive. Yet, deep down, for those willing to be honest, there is something not fully healthy in it.” --Francois Pinard R

90
Thank you! May the R be with you!

91
Todo multiple panels lists, loops, lapply, sapply regular expressions

Similar presentations

>")






![Recitation 4. 2-D arrays. Exceptions. Animal[] v= new Animal[3]; 2 declaration of array v v null Create array of 3 elements a6 Animal[] 012012 null Assign.](/8/2339022/big_thumb.jpg "Recitation 4. 2-D arrays. Exceptions. Animal[] v= new Animal[3]; 2 declaration of array v v null Create array of 3 elements a6 Animal[] 012012 null Assign.>")








>")

