Download presentation
Presentation is loading. Please wait.

1
Relaxation and Dynamics, and advanced methods
Chem 781 Part 9 Relaxation and Dynamics, and advanced methods

2
NOE is a relaxation phenomenon
Measure of dipolar interaction between two spins NOEs can not be observed between two equivalent nuclei. Direct measurement of relaxation time allows for measurement of dipolar coupling between equivalent spins

3
Measurement of longitudinal relaxation time T1
Inversion recovery The experiment is repeated with values of T between 0 and 5 T1

4
Relaxation of longitudinal magnetization
By definition, longitudinal relaxation is the recovery of z magnetization towards equilibrium. For two spins IA and IB coupled by dipolar coupling it is dIzA/dt = (2 W1A + W0 +W2) (I0A – IzA) + (W2-W0) (I0B – IzB) 9.1 If IA ≠ IB (for example 13C-1H) and we decouple IB (1H), then longitudinal relaxation depends only on the first term and IzB = constant = 0 throughout the experiment. As already mentioned in part one of the lecture we get an mono exponential recovery of z-magnetization: The above equation assumes Mz(0) = 0 (90° pulse or saturation). The second term in 9.1 only influences the equilibrium value M0 by accounting for the NOE.
 (I0A – IzA) + (W2-W0) (I0B – IzB) 9.1. If IA ≠ IB (for example 13C-1H) and we decouple IB (1H), then longitudinal relaxation depends only on the first term and IzB = constant = 0 throughout the experiment. As already mentioned in part one of the lecture we get an mono exponential recovery of z-magnetization: The above equation assumes Mz(0) = 0 (90° pulse or saturation). The second term in 9.1 only influences the equilibrium value M0 by accounting for the NOE.")
5
T1 recovery curve Obtain T1 by fitting recovery curve to exponential equation with Mz(0) = - Mz0 :
 = - Mz0 :")
6
Longitudinal Relaxation by dipolar coupling
Using the relations for W1, W0 and W2 used for NOE, one obtains for non-equivalent spins from 9.1: 1/T1A = (2W1A + W0AB + W2) = 1/10D2AB[J(ωA-ωB) + 3J(ωA) + 6J(ωA+ωB)] (heteronuclear) For equivalent spins (IA = IB) one obtains from 9.1 with IzB = IzA 1/T1A = (2W1A+2W2AB) = 3/10 D2AB [J(ωA) + 4J(2ωA)] (homonuclear) The expressions depend both on the dipolar coupling and the motion of the molecule: DAB = (γAγB ℏ2)/rAB contains the distance information Contains the motion of the molecule
 = 1/10D2AB[J(ωA-ωB) + 3J(ωA) + 6J(ωA+ωB)] (heteronuclear) For equivalent spins (IA = IB) one obtains from 9.1 with IzB = IzA. 1/T1A = (2W1A+2W2AB) = 3/10 D2AB [J(ωA) + 4J(2ωA)] (homonuclear) The expressions depend both on the dipolar coupling and the motion of the molecule: DAB = (γAγB ℏ2)/rAB3 contains the distance information. Contains the motion of the molecule.")
7
Longitudinal relaxation (T1) and distance
To measure distance using T1 we need to know motion One could try to measure motion (τc) independently, but that is typically not easily possible Relaxation will be most efficient when . This point can be often obtained by changing the temperature and finding the minimum T1 (the correlation time will depend on temperature) T1(min) only depends on the distance and the field:
 independently, but that is typically not easily possible. Relaxation will be most efficient when . This point can be often obtained by changing the temperature and finding the minimum T1 (the correlation time will depend on temperature) T1(min) only depends on the distance and the field:")
8
Example: Dihydride vs. dihydrogen complex
T1(min) of 0.23 s is only compatible with dihydride structure. However, the value can not be used to calculate the exact bond distance, since the equation assumed an isolated pair of protons. There are other protons around, and other relaxation mechanisms possible.
 of 0.23 s is only compatible with dihydride structure. However, the value can not be used to calculate the exact bond distance, since the equation assumed an isolated pair of protons. There are other protons around, and other relaxation mechanisms possible.")
9
Schematic Spin-Lattice vs. Spin-Spin relaxation

10
Relaxation of x,y magnetization (transverse relaxation)
Dephasing of x,y magnetization is caused by both random transitions between levels (longitudinal relaxation) AND incomplete averaging of orientation dependent shifts (dephasing by chemical exchange) That results in an additional J(0) term 1/T2A = 1/10D2AB [4 J(0) + J(ωA-ωB) + 3J(ωA) + 6J(ωA+ωB)] Spin-spin Spin-Lattice (Same as T1)
 AND incomplete averaging of orientation dependent shifts (dephasing by chemical exchange) That results in an additional J(0) term. 1/T2A = 1/10D2AB [4 J(0) + J(ωA-ωB) + 3J(ωA) + 6J(ωA+ωB)] Spin-spin. Spin-Lattice (Same as T1)")
11
Fast tumbling averages dipolar coupling to zero

12
Measurement of T2 Spin echo experiment
We need to separate the effects of inhomogeneity (reversible) from relaxation of x,y magnetization (irreversible): Spin echo experiment Take a series of experiments and vary the number of 180⁰ pulses with refocusing delay (n). The intensity of the resulting spectrum will decay with T2 : Mx,y = exp(-2nτ/T2)
 from relaxation of x,y magnetization (irreversible): Spin echo experiment. Take a series of experiments and vary the number of 180⁰ pulses with refocusing delay (n). The intensity of the resulting spectrum will decay with T2 : Mx,y = exp(-2nτ/T2)")
13
T1 and T2 vs. correlation time
For short correlation times (small molecules), T1 = T2 For long correlation times,(large molecules), T2 is getting shorter and shorter => Lines are getting broader as molecule gets larger
, T1 = T2. For long correlation times,(large molecules), T2 is getting shorter and shorter. => Lines are getting broader as molecule gets larger.")
14
Relaxation Mechanisms
Relaxation can occur through many mechanisms: While cross relaxation (and thus NOE) is solely determined by dipolar coupling, other mechanisms can contribute additive to the overall longitudinal relaxation rate 1/T1total = 1/T1dipol + 1/T1CSA + 1/T1Quad + 1/T1SR + 1/T1paramagnetic ... CSA: Chemical shift anisotropy Quad: Quadrupol coupling SR: Spin rotation For protons and C-H, N-H usually dipolar coupling is dominant, but for non protonated carbons or quadrupolar nuclei other relaxation mechanisms become important. In paramagnetic molecules, the electron-nucleus interaction is often dominant.
 is solely determined by dipolar coupling, other mechanisms can contribute additive to the overall longitudinal relaxation rate. 1/T1total = 1/T1dipol + 1/T1CSA + 1/T1Quad + 1/T1SR + 1/T1paramagnetic ... CSA: Chemical shift anisotropy Quad: Quadrupol coupling SR: Spin rotation. For protons and C-H, N-H usually dipolar coupling is dominant, but for non protonated carbons or quadrupolar nuclei other relaxation mechanisms become important. In paramagnetic molecules, the electron-nucleus interaction is often dominant.")
15
Relaxation by chemical shift anisotropy:
Chemical shielding can depend on the orientation of the molecule with respect to the field. While in solution only an average isotropic shift is observed, the nucleus actually experiences an fluctuating local field. For axial symmetry the magnitude of the field fluctuation is given by the difference between the orientations with maximum and minimum shielding (σ∥ and σ⊥):
:")
16
Importance of CSA CSA is an important relaxation mechanism for tertiary C=O and C≡ groups as there are no protons nearby and the anisotropy of the shielding is particulary large. CSA is also important for heavy I = ½ nuclei (103Rh, 183W, ...) As they also exhibit large chemical shift ranges. Note the dependence on B02 for 1/T1CSA. At higher fields this relaxation mechanism becomes more important. That is good news for metal NMR as repetition times are reduced, but may cause less than maximum NOE’s for C-H groups at very high field. Measurement of T1 at different fields allows to separate T1CSA from T1DD and other relaxation
 As they also exhibit large chemical shift ranges. Note the dependence on B02 for 1/T1CSA. At higher fields this relaxation mechanism becomes more important. That is good news for metal NMR as repetition times are reduced, but may cause less than maximum NOE’s for C-H groups at very high field. Measurement of T1 at different fields allows to separate T1CSA from T1DD and other relaxation.")
17
Relaxation from Quadrupol interaction
This mechanism occurs only for I > ½ as those nuclei are not spherical but shaped like an ellipsoid In a non symmetric environment different orientations of the nucleus relative to the environment will have different energies (interaction with electric field gradient). As this energy can be quite large (several MHz) it is the dominant relaxation mechanism for all I > ½ nuclei and results in often extremely short relaxation times for these nuclei.
. As this energy can be quite large (several MHz) it. is the dominant relaxation mechanism for all. I > ½ nuclei and results in often extremely. short relaxation times for these nuclei.")
18
Consequences of quadrupolar relaxation
I > ½ nuclei usually have very broad lines except when in very symmetric environments (octahedral or tetrahedral) and/or for nuclei with very small quadrupolar coupling constant (2D, 11B, 7Li) A very small repetition delay d1 (0.1s) and acquisition time (td = 4k) can be used in many cases almost never is NOE observed for any of those nuclei (7Li NMR is one exception) For neighboring nuclei the fast relaxation acts like decoupling (if 1/T1 > J) and couplings are often not observed: that is why we don’t see 14N-H coupling (14N > 99%). Only very large couplings or couplings to low QC nuclei is observed, or in case of a very symmetrical molecule like NH4+. In some cases, neighboring coupling partners will appear broadened: one other reason why N-H protons might appear broadened
 and/or for nuclei with very small quadrupolar coupling constant (2D, 11B, 7Li) A very small repetition delay d1 (0.1s) and acquisition time (td = 4k) can be used in many cases. almost never is NOE observed for any of those nuclei (7Li NMR is one exception) For neighboring nuclei the fast relaxation acts like decoupling (if 1/T1 > J) and couplings are often not observed: that is why we don’t see 14N-H coupling (14N > 99%). Only very large couplings or couplings to low QC nuclei is observed, or in case of a very symmetrical molecule like NH4+. In some cases, neighboring coupling partners will appear broadened: one other reason why N-H protons might appear broadened.")
19
Other relaxation mechanisms
Interaction with unpaired electrons (spin) will be discussed with NMR of paramagnetic compounds. Note that O2 is paramagnetic and best NOE results of small molecules require degassing of sample. Spin Rotation: currents induced by rotation of molecule, only important for very small molecules and in gas phase, sometimes methyl groups
 will be discussed with NMR of paramagnetic compounds. Note that O2 is paramagnetic and best NOE results of small molecules require degassing of sample. Spin Rotation: currents induced by rotation of molecule, only important for very small molecules and in gas phase, sometimes methyl groups.")
20
Problem of measuring ultra large molecules
T2 relaxation time becomes shorter as Molecule becomes bigger Increasing line width not only causes more overlap, but also at some point makes magnetization transfer impossible Typically, different relaxation mechanisms are additive. However, as the motions leading to different relaxation mechanisms are the same, the effects can sometimes cancel or subtract

21
Motions modulating the different mechanisms are not independent
1/T1total = 1/T1dipol + 1/T1CSA + 1/T1Quad + 1/T1SR + 1/T1paramagnetic ... The different relaxation rates will only add up if the motions are independent for the different mechanisms Since often different interactions are modulated by the same motion, the different relaxation mechanisms are often not independent Cumulative effects of different relaxation mechanisms are not straightforward

22
Example: Dipolar- and CSA relaxation of N-15

23
Consequences of correlated relaxation
Nitrogen N-Ha line will relax faster than the N-Hb line For large molecules, T2 for the N-Ha line will be shorter and the line will be broader, and the N-Hb line will be sharper At high enough magnetic field magnitude of CSA will equal dipolar coupling, and the two interactions will cancel for N-Hb line Hb Hb n0N

24
For a peptide N-H nitrogen, CSA and dipolar relaxation cancel for one doublet line, and add for the other In a regular N-15 decoupled HSQC the broader line would still dominate the linewidth of the spectrum Instead it is better to take an experiment that observes only the sharp line and discards the other. => Transverse Relaxation Optimized Spectroscopy (TROSY)
")
25
Small molecule example of TROSY:
Use viscous solvent and low temperature to achieve short T2

26
2D TROSY selects

27
Large Molecule application of TROSY
2D 1H-15N HSQC spectrum of the same 35 kDa tumor suppressor protein 2D 1H-15N TROSY spectrum of a 35 kDa tumor suppressor protein at 900 MHz. Rubin group, UC Santa Cruz

28
NMR and dynamics

29
Relaxation and molecular dynamics
If the magnitude of the interaction (i.e. distance) is known, the relaxation times can be used to probe for molecular motion Example 13C-H bonds: Due to its low natural abundance, and the r-6 dependence on the distance, relaxation of the 13C nucleus of a C-H group usually solely depends on dipolar relaxation to the directly attached hydrogen. As there are many data on C-H bond lengths, 13C relaxation measurements can be used to probe for molecular motion For small molecules the relaxation time is proportional to the correlation time::
 is known, the relaxation times can be used to probe for molecular motion. Example 13C-H bonds: Due to its low natural abundance, and the r-6 dependence on the distance, relaxation of the 13C nucleus of a C-H group usually solely depends on dipolar relaxation to the directly attached hydrogen. As there are many data on C-H bond lengths, 13C relaxation measurements can be used to probe for molecular motion. For small molecules the relaxation time is proportional to the correlation time::")
30
Example aliphatic chains
The above equation is valid for a rigid spherical molecule. In real molecules, fast internal rotation about single bonds will contribute to the diffusion coefficient, resulting in different correlation times for rigid and flexible parts of the molecule. Relaxation measurements thus can reveal internal rotations in the MHz range.

31
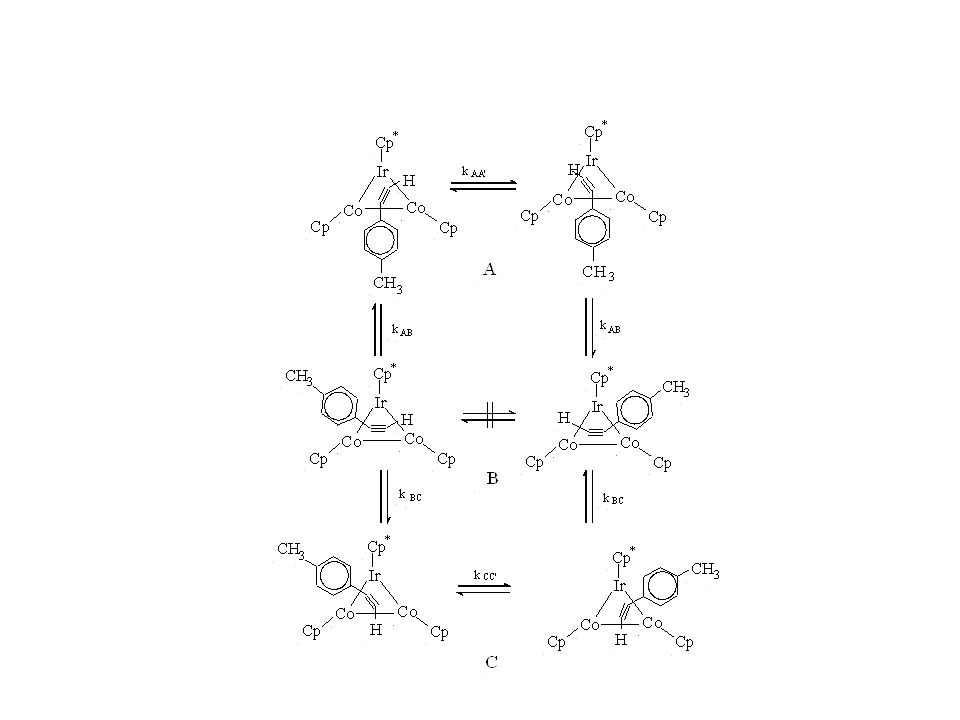
Anisotropic and internal rotation
Often the overall tumbling of the molecule is anisotropic, i.e. tumbling about different axes takes place at different rates. Bistolane and the shown cobalt cluster complex are cigar shaped and the relaxation of the phenyl carbons depends on three diffusion coefficients: tumbling perpendicular to the main axis (D⊥), tumbling parallel to the main axis (D∥) and internal rotation RPh. Quantitative analysis of such data can become very tedious:
, tumbling parallel to the main axis (D∥) and internal rotation RPh. Quantitative analysis of such data can become very tedious:")
32
where A = (3 cos2Θ-1)/4, B = 3 sin2Θ cos2Θ and C = (3 sin4Θ)/4 with Θ the angle between the C-H bond and the rotational axis. For Cpara the C-H bond is part of the rotation axis for both internal rotation and D∥. Dipolar coupling for this for this group is thus only modulated by D⊥ which is considerably smaller and thus relaxation times for these carbons are much shorter.

33
Backbone dynamics of proteins
Relaxation data reveal increased flexibility in the linker region. Also, the difference in correlation time of the two domains can be related to the slightly different sizes of the domains.

34
Dynamics from Lineshape
Relaxation is sensitive for dynamics in the scale of MHz Dynamics has an effect on the the line shape of a signal when interchange between two groups of different chemical shift becomes of the same order of magnitude as the chemical shift separation measured in Hz Typically 1 s-1 – 10-3 s-1 Much faster interchange gives one average signal Much slower exchange gives two separate signals

35
Example N,N-dimethylformamide
In the intermediate temperature range the two peaks will broaden with rising temperature, merge into one and then gradually sharpen to give one sharp signal. The temperature where the signal is at its broadest is called the coalescence point The rate at the coalescence point is given by From: “Measuring Rates by NMR”, Hans Reich,

36
Approximations to measure rates
Finding the exact coalescence temperature is not always trivial or practical At temperatures below the coalescence point (slow exchange limit) the line width can be used to approximately determine kAB. kAB = π Δνex (Slow exchange limit) The exchange broadening Δνex is obtained from the line width by subtracting the natural line width Δνex = Δνobs - Δν0 at temperatures above the coalescence point: kAB = π(νA-νB)2/(2Δνex) (Fast exchange limit) Note the need to determine or at least estimate the chemical shift difference
 the line width can be used to approximately determine kAB. kAB = π Δνex (Slow exchange limit) The exchange broadening Δνex is obtained from the line width by. subtracting the natural line width Δνex = Δνobs - Δν0. at temperatures above the coalescence point: kAB = π(νA-νB)2/(2Δνex) (Fast exchange limit) Note the need to determine or at least estimate the chemical shift difference.")
37
Line shape analysis exchange can involve more than two species
Intermolecular exchange can involve species of different populations (equilibrium constant KC) => the broadening pattern can become very complex and the above approximations will not be applicable. Full simulation of line shape possible with a computer to determine rate constants (line shape analysis) becomes necessary This allows the rate constant to be determined for multiple temperatures
 => the broadening pattern can become very complex and the above approximations will not be applicable. Full simulation of line shape possible with a computer to determine rate constants (line shape analysis) becomes necessary. This allows the rate constant to be determined for multiple temperatures.")
40
Experimental simulated

41
Exchange and magnetization transfer
Chemical exchange which is slow on the chemical shift time scale can have a similar effect on NOE spectra as relaxation via W0 . In order to be effective two conditions have to be fulfilled: one has to observe two separate signals, which means kAB << ΔδAB the rate of exchange needs to be larger or at least not much slower than longitudinal relaxation rate The condition for magnetization transfer via exchange is (νA-νB) > kAB > 1/T1 Exchange too slow to give broadening still can still be probed by NMR using magnetization transfer. Usually that is the case for rate constants of the order 10-2 s s-1
 > kAB > 1/T1. Exchange too slow to give broadening still can still be probed by NMR using magnetization transfer. Usually that is the case for rate constants of the order 10-2 s s-1.")
42
1D Magnetization transfer
14.9 ppm -25.4 ppm

43
Numerical best parameter fit

44
2D NOESY techniques:

45
Fitting NOE cross peak intensities

46
Rate constants and activation barriers
Measuring the temperature dependence of rate constants yields activation barrier Eyring equation: kAB : rate constant of exchange k : Boltzmann constant Typically a logarithmic plot is obtained: The accuracy in particular of ΔS≠ depends strongly on the temperature range sampled.

47
Sensitivity of NMR Experiments
Signal/Noise ∼ N∙ 𝑛𝑠 ∙ Polarization ∙ μobserved ∙ induction ∙ T2*/T1 ∙QProbe ∙ Efficiency γB0/kT γ 𝐼(𝐼+1) ∼ ω0 = γB0 Higher magnetic field (20 T currently max.) $$$$ Reduce noise of electronics: cool detection circuit and preamplifier with cold heluim gas (cryo probe) or nitrorogen gas (cryo probe Prodigy) $$ Concentrate sample and scale down dimension of probe ( micro probe)
 ∼ ω0 = γB0. Higher magnetic field (20 T currently max.) $$$$ Reduce noise of electronics: cool detection circuit and preamplifier with cold heluim gas (cryo probe) or nitrorogen gas (cryo probe Prodigy) $$ Concentrate sample and scale down dimension of probe ( micro probe)")
48
Improving sensitivity: manipulate Boltzmann distribution
In some cases, coupling to higher energy levels (rotational, optical, electron spin) can be used to obtain highly improved population differences Dynamic Nuclear Polarization (DNP) Chemically induced nuclear polarization (CIDNP) Para hydrogen induced Nuclear Polarization All methods are currently commercialized for more general use
 can be used to obtain highly improved population differences. Dynamic Nuclear Polarization (DNP) Chemically induced nuclear polarization (CIDNP) Para hydrogen induced Nuclear Polarization. All methods are currently commercialized for more general use.")
49
Dihydrogen Gas is a mixture of two Spin Isomers
Singlet Para-hydrogen Triplet Ortho-hydrogen ab-ba S (singlet) I =0, m =0 tot s T (triplet) =1 m aa +1 ab+ba bb -1 J Singlet-Triplet conversion forbidden, and each isomer is stable in pure hydrogen gas At room temperature: 75% orthohydrogen (expected from Boltzmann distribution) At 77 K (Liq. N2): % parahydrogen NOT expected by a simple splitting 20 K > 99% parahydrogen of levels by J = 240 Hz
 I. =0, m. =0. tot. s. T (triplet) =1. m. aa. +1. ab+ba. bb. -1. J. Singlet-Triplet conversion forbidden, and each isomer is stable in pure hydrogen gas. At room temperature: 75% orthohydrogen (expected from Boltzmann distribution) At 77 K (Liq. N2): 50 % parahydrogen NOT expected by a simple splitting. 20 K > 99% parahydrogen of levels by J = 240 Hz.")
50
Origin of the high energy difference
ΨDihydrogen = ψtrans • ψvibr • ψrot • ψspin The whole nuclear wave function needs to be considered General Pauli Principle: The total wave function of spin ½ particles is always anti symmetric with respect to exchange of two particles Ψ(1,2) = - Ψ(2,1) Translation only depends on center of gravity } Always Symmetrical Vibration only depends on absolute distance Rotation levels can be symmetrical for even quantum numbers 0,2,4,… (s,d,…) or anti symmetrical for odd quantum numbers 1,3,5,… (p,f, …) Singlet spin function is anti symmetric, triplet spin function is symmetric symmetric rotational function can only combine with anti symmetric spin function and vice versa
 = - Ψ(2,1) Translation only depends on center of gravity. } Always Symmetrical. Vibration only depends on absolute distance. Rotation levels can be symmetrical for even quantum numbers 0,2,4,… (s,d,…) or anti symmetrical for odd quantum numbers 1,3,5,… (p,f, …) Singlet spin function is anti symmetric, triplet spin function is symmetric. symmetric rotational function can only combine with anti symmetric spin function and vice versa.")
51
Population difference of spin states determined by Rotational states
Only combination of symmetrical rotational state and anti-symmetrical spin function OR anti symmetrical rotational state with symmetric spin function are allowed Population difference will be given by rotational energy, several orders of magnitude larger than magnetic interaction In absence of a catalyst, there will be no inter-conversion of triplet to singlet state Also, no transitions are allowed between S and T

52
ΨS(1,2) = ϕ1(1) ϕ 2(2) + ϕ1(2) ϕ2(1) symmetric
Analogy to Hund’s rule:Two electrons in two degeneratre orbitals ϕ1 and ϕ2: Two possible wave functions , one symmetric and one anti symmetric: ΨS(1,2) = ϕ1(1) ϕ 2(2) + ϕ1(2) ϕ2(1) symmetric ΨA(1,2) = ϕ1(1) ϕ 2(2) - ϕ1(2) ϕ2(1) anti-symmetric Energies of ΨA and ΨS will be different, with ΨA lower in energy due to electron repulsion As total wave function needs to be anti-symmetric, ΨA will only go with symmetric spin function, and Ψ S only with anti symmetric spin function Also note that if ϕ1= ϕ2 (two electrons in the same orbital), ΨA will be zero and only Ψ S will exist. ΨS(1,2) ΨA(1,2) ∆E determined by difference in electron wave function (coulomb e-- e- repulsion)
 = ϕ1(1) ϕ 2(2) + ϕ1(2) ϕ2(1) symmetric. ΨA(1,2) = ϕ1(1) ϕ 2(2) - ϕ1(2) ϕ2(1) anti-symmetric. Energies of ΨA and ΨS will be different, with ΨA lower in energy due to electron repulsion. As total wave function needs to be anti-symmetric, ΨA will only go with symmetric spin function, and Ψ S only with anti symmetric spin function. Also note that if ϕ1= ϕ2 (two electrons in the same orbital), ΨA will be zero and only Ψ S will exist. ΨS(1,2) ΨA(1,2) ∆E determined by difference in electron wave function (coulomb e-- e- repulsion)")
53
How does that help with NMR ?
To be useful for NMR, two conditions need to be met: conversion ortho – para needs to be fast to enrich para-H2 in reasonable time After enrichment, symmetry needs to be broken fast enough to observe transitions between former T to S states while maintaining polarization

54
Presence of metal catalysts to speed up para to ortho equilibrium
Ortho and Para hydrogen usually do not interconvert Temporary breaking of H-H bond will allow equilibrium to be achieved Any metal that weakly binds hydrogen will do Frozen solutions of hydrogenation catalysts stored under hydrogen gas at liquid nitrogen temperature will do, for example inside NMR tube

55
Hydrogenation reaction breaks symmetry of dihydrogen molecule

56
Kirill V. Kovtunov a, Vladimir V
Kirill V. Kovtunov a, Vladimir V. Zhivonitko a, Lioubov Kiwi-Minsker b and Igor V. Koptyug *aChem. Commun., 2010, 46,

57
Paramagnetic Molecules
Molecules with unpaired electrons have net electron spin Electron paramagnetic resonance same principle as NMR Negative γ from negative charge results in -1/2 (anti parallel) state lower in energy Similar parameters: g- value and hyperfine coupling (electron-nucleus scalar coupling)
 state lower in energy. Similar parameters: g- value and hyperfine. coupling (electron-nucleus scalar coupling)")
58
EPR spectra typically displayed in dispersion mode
H2C(OCH3) radical
 radical.")
59
NMR and EPR complimentary
EPR is possible if electron relaxation is slow enough. NMR is typically not possible in those molecules If electron relaxation is fast, EPR becomes difficult, but NMR spectra are observable Typically in bi-radicals or metal complexes

60
Example transition metal complexes
Lines typically broad and extremely shifted Shift can be positive or negative Cp2Co 19 electrons δ(H) = ppm 46 electrons δ(H) = - 30 ppm
 = ppm. 46 electrons δ(H) = - 30 ppm.")
62
Origin of extreme chemical shift
Shift in paramagnetic complexes arises from scalar coupling to electron (contact shift) Coupling is extremely large (MHz), so the two lines will not be equally populated Slow electron relaxation will effectively wipe out signal (but EPR is possible) Fast electron relaxation will give average signal that is shifted towards the higher populated line Depending on sign of coupling constant shift is positive or negative
 Coupling is extremely large (MHz), so the two lines will not be equally populated. Slow electron relaxation will effectively wipe out signal (but EPR is possible) Fast electron relaxation will give average signal that is shifted towards the higher populated line. Depending on sign of coupling constant shift is positive or negative.")
63
Application: Shift Reagent

64
Metallo- Protein and Metal DNA complexes

65
Multinuclear NMR

Similar presentations






 (Fall Term, 2005) Department of Chemistry National Sun Yat-sen University 無機物理方法(核磁共振部分)>")


 Spectroscopy>")
>")
>")
>")





 A photon generates both an electric and a magnetic field A current passing through a wire also generates both an electric and a magnetic.>")


 FID represents the time-domain response of the spin system following application.>")
>")

