Download presentation
Presentation is loading. Please wait.

1
ABC: Bayesian Computation Without Likelihoods David Balding Centre for Biostatistics Imperial College London (www.icbiostatistics.org.uk)

2
Bayesian inference via rejection from prior I Generate a posterior random sample for a parameter of interest θ by a mechanical version of Bayes Theorem: 1.simulate θ from its prior; 2.accept/reject, with P(accept) likelihood; 3.if not enough acceptances yet, go to 1. Problem: if likelihood involves integration over many nuisance parameters, hard/slow to compute. Solution: use simulation to approximate likelihood.

3
Bayesian inference via rejection from prior II Generate an approximate posterior random sample: 1.simulate parameter vector θ from its prior; 2.simulate data X given value of θ from 1.; 2a.if X matches observed data, accept θ; 3.if not enough acceptances yet, go to 1. Problem: simulated X hardly ever matches observed. Solution: relax 2a so that θ is accepted when X is close to observed data; close to is usually measured in terms of a vector of summary statistics, S.

4
Summary statistic, S Parameter, Prior – p( ) Marginal likelihood – p(S) Posterior density – p( | S) Likelihood – p(S | )
 Marginal likelihood – p(S) Posterior density – p( | S) Likelihood – p(S | )")
5
Approximate Bayesian Computing (ABC) We simulate to approximate (1) the joint parameter/ data density then (2) a slice at the observed data. Few if any simulated points will lie on this slice so need to assume smoothness: required posterior is approximately the same for datasets close to that observed. Note: (1) we get approximate likelihood inferences but we didnt calculate the likelihood (2) different definitions of close can be tried for the same set of simulations (3) these can even be retained and used for different observed datasets.
 we get approximate likelihood inferences but we didnt calculate the likelihood (2) different definitions of close can be tried for the same set of simulations (3) these can even be retained and used for different observed datasets..")
6
θ values of these points are treated as random sample from posterior

8
When to use ABC ? When likelihood is hard to compute because of need for integration over many nuisance parameters BUT easy to simulate –Population genetics: nuisance parameters are the branching times and topology of the genealogical tree underlying the observed DNA sequences/genes. –Epidemic models: nuisance parameters are infection times and infectious periods. ABC implies 3 approximations: 1. finite # simulations; 2. non-sufficiency of S; 3. S need not match S exactly

9
Population genetics example Parameters: N = effective population size; μ = mutation rate per generation; G = genealogical tree (topology + branch lengths) – nuisance Summary Statistics: S 1 number of distinct alleles/sequences S 2 number of polymorphic/segregating sites Algorithm: 1. simulate N and μ from joint prior 2. simulate G from the standard coalescent model 3. simulate mutations on G and calculate S* 4. accept (N, μ,G) if S* S This generates a sample from the joint posterior of (N, μ,G). To make inference about θ =2Nμ, simply ignore G.
 if S* S This generates a sample from the joint posterior of (N, μ,G). To make inference about θ =2Nμ, simply ignore G..")
10
Model comparison via ABC Can also use ABC for model comparison, as well as for parameter estimation within models. Ratio of acceptances: approximates the Bayes Factor. Better: fit (weighted) multinomial regression to predict model from observed data. Beaumont (2006) used this to infer the topology of a tree representing the history of 3 Californian fox populations.
 multinomial regression to predict model from observed data. Beaumont (2006) used this to infer the topology of a tree representing the history of 3 Californian fox populations..")
11
Problems/limitations Rejection-ABC is very inefficient: most simulated datasets are far from observed and must be rejected. No learning. How to find/assess good summary statistics? –Too many summary statistics can make matters worse (see later) How to choose metric for (high-dimensional) S
 How to choose metric for (high-dimensional) S.")
12
Beaumont, Zhang, and DJB Approximate Bayesian Computation in Population Genetics. Genetics 162: 2025-2035, 2002 Use local-linear regression to adjust for the distance between observed and simulated datasets. Use a smooth (Epanechnikov) weighting according to distance. Can now weaken the close criterion (i.e. increase the tolerance) and utilize many more points.
 weighting according to distance. Can now weaken the close criterion (i.e. increase the tolerance) and utilize many more points..")
13
1 0 Summary Statistic Weight Parameter

14
1 0

19
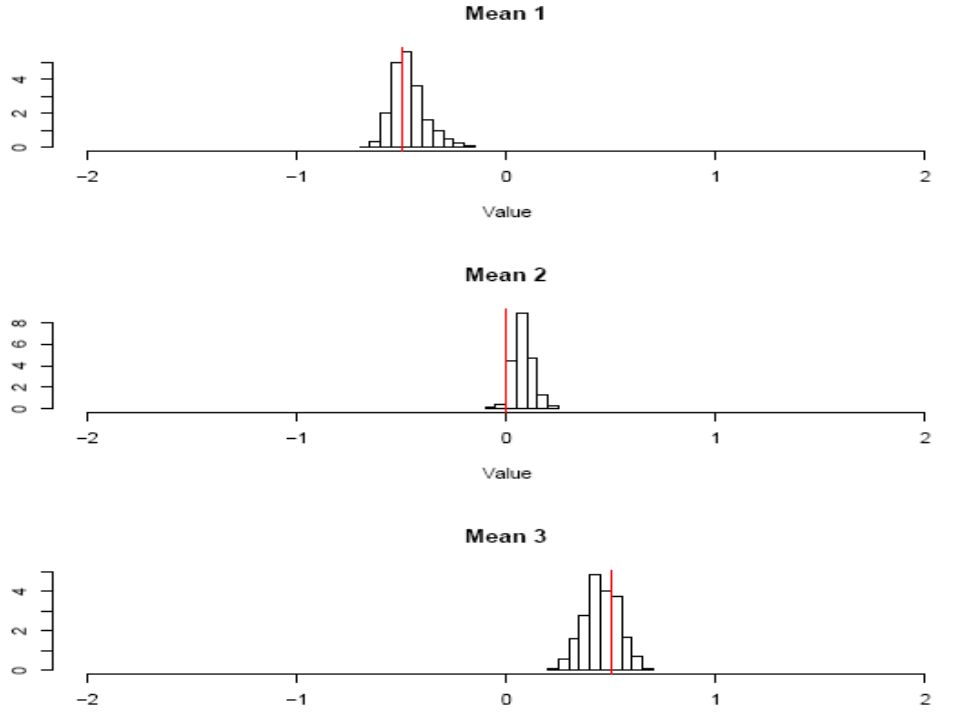
Estimation of scaled mutation rate = 2N Tolerance Relative mean square error MCMC Standard Rejection With regression adjustment Summary statistics:- mean variance in length mean heterozygosity number of haplotypes i.e. 3 numbers Full data:- 445 Y chromosomes each typed at 8 microsatellite loci i.e. 3560 numbers

20
Population growth Population constant size N A until t generations ago, then exponentially rate r per gen. growth to N C. 4 model params, but only 3 identifiable. We choose: Data same as above, except smaller sample size n = 200 (because of time taken for MCMC to converge).
..")
23
Standard rejection method : Estoup et al. (2002, Genetics)– Demographic history of invasion of islands by cane toads. 10 microsatellite loci, 22 allozyme loci. 4/3 summary statistics, 6 demographic parameters. Estoup and Clegg (2003, Molecular Ecology) – Demographic history of colonisation of islands by silvereyes. With regression adjustment : Tallmon et al (2004, Genetics) – Estimating effective population size by temporal method. One main parameter of interest (Ne), 4 summary statistics. Estoup et al. (2004, Evolution) – Demographic history of invasion of Australia by cane toads. 75/63 summary statistics, model comparison, up to 5 demographic parameters. ABC applications in population genetics:
– Demographic history of invasion of islands by cane toads. 10 microsatellite loci, 22 allozyme loci. 4/3 summary statistics, 6 demographic parameters. Estoup and Clegg (2003, Molecular Ecology) – Demographic history of colonisation of islands by silvereyes. With regression adjustment : Tallmon et al (2004, Genetics) – Estimating effective population size by temporal method. One main parameter of interest (Ne), 4 summary statistics. Estoup et al. (2004, Evolution) – Demographic history of invasion of Australia by cane toads. 75/63 summary statistics, model comparison, up to 5 demographic parameters. ABC applications in population genetics:.")
24
More sophisticated regressions? Although global linear regression usually gives a poor fit to joint θ/S density, Calabrese (USC, unpublished) uses projection pursuit regression: to fit a large feature set of summary statistics. Iterate to improve fit within vicinity of S. Application to estimate human recombination hotspots. Could also consider quantile regression to adapt adjustment to different parts of the distribution.
 uses projection pursuit regression: to fit a large feature set of summary statistics. Iterate to improve fit within vicinity of S. Application to estimate human recombination hotspots. Could also consider quantile regression to adapt adjustment to different parts of the distribution..")
25
Do ABC within MCMC Marjoram et al. (2003). Two accept/reject steps: 1.Simulate a dataset at the current parameter values; if it isnt close to observed data, start again. 2.If it is close, accept or reject according to prior ratio times Hastings ratio (no likelihood ratio) Note: now close must be defined in advance; also cannot reuse simulations for different observed datasets. Can apply regression- adjustment to MCMC outputs. Problems: 1.proposals in tree space 2.few acceptances in tail of target distribution - stickiness
 Note: now close must be defined in advance; also cannot reuse simulations for different observed datasets. Can apply regression- adjustment to MCMC outputs. Problems: 1.proposals in tree space 2.few acceptances in tail of target distribution - stickiness.")
26
Importance sampling within MCMC In fact, the Marjoram et al. MCMC approach can be viewed as a special case of a more general approach developed by Beaumont (2003). Instead of simulating a new dataset forward-in-time, Beaumont used a backward-in-time IS approach to approximate the likelihood. His proof of the validity of the algorithm is readily extended to forwards-in-time approaches based on one or multiple datasets (cf ONeill et al. 2000). Could also use a regression adjustment.
. Instead of simulating a new dataset forward-in-time, Beaumont used a backward-in-time IS approach to approximate the likelihood. His proof of the validity of the algorithm is readily extended to forwards-in-time approaches based on one or multiple datasets (cf ONeill et al. 2000). Could also use a regression adjustment..")
27
ABC within Sequential MC Sisson et al at UNSW, Sydney Sample initial generation of θ particles from prior. Sample θ from previous generation, propose new value and generate dataset; calculate S*. Repeat until S* S – BUT tolerance reduces each gen. Calculate prior ratio times Hastings ratio: use as weight W for sampling the next generation. If variance of W is large, resample with replacement according to W and set all W=1/N. Application to estimate parameters of TB infection.

28
Adaptive simulation algorithm (Molitor and Welch, in progress) simulate N values of θ from prior calculate corresponding datasets and use similarity of S* with S to generate a density resample from density, replace value with lowest similarity of S* and S. use final density as importance sampling weights for a conventional ABC. –idea is to use preliminary pseudo-posterior based on weights to choose something better than prior as basis for ABC

35
"number of data generation steps for rejection ABC" [1] 35064[2] 27877 "number of data generation steps for SMC ABC" [1] 14730[2] 12629 "number of data generation steps for Johns ABC" [1] 10314[2] 6130
![number of data generation steps for rejection ABC [1] 35064[2] number of data generation steps for SMC ABC [1] 14730[2] number of data generation steps for Johns ABC [1] 10314[2] 6130](http://images.slideplayer.com/3/781255/slides/slide_35.jpg "number of data generation steps for rejection ABC [1] 35064[2] number of data generation steps for SMC ABC [1] 14730[2] number of data generation steps for Johns ABC [1] 10314[2] 6130")
36
ABC to rescue poor estimators (inspired by DJ Wilson, Lancaster) evaluate estimator based on simplistic model at many datasets simulated under more sophisticated model. for observed dataset, use as estimator regression predictor of simplistic estimator at the observed data value. for example, many population genetics estimators assume no recombination, and infinite sites mutation model –use this estimator and simulations to correct for recombination and finite-sites mutation

37
Acknowledgments David Welch and John Molitor, both of Imperial College. David has just started on an EPSRC grant to further develop ABC ideas and apply particularly in population genomics.

Similar presentations













 methods>")


 Dpt. of System Engineering and Automation May 19-23 Pasadena,>")







 is sufficient for >")


>")

