Download presentation
Presentation is loading. Please wait.

1
Human Gene Mapping & Disease Gene Identification

2
Overview This chapter provides an overview of how geneticists use the familial nature of disease to identify the responsible genes and gene variants. Whether a disease is inherited in a recognizable mendelian pattern or just occurs at a higher frequency in relatives of affected individuals, the genetic contribution to disease must result from genotypic differences among family members that either cause disease outright or increase or decrease disease susceptibility.

3
The Human Genome Project, has provided geneticists with a complete list of all human genes, knowledge of their location and structure, and a catalogue of some of the millions of variants in DNA sequence found among individuals in different populations. Some of these variants are common, others are rare, and still others differ in frequency among different ethnic groups. Whereas some variants clearly have functional consequences, others are certainly neutral. For most, their significance for human health and disease is unknown.

4
In this chapter we discuss how meiosis, acting over both time and space, determines the relationships b/w genes and polymorphic loci with their neighbors Two fundamental approaches to disease gene identification: linkage analysis, is family-based. Linkage analysis takes explicit advantage of family pedigrees to follow the inheritance of a disease over a few generations by looking for consistent, repeated inheritance of a particular region of the genome whenever disease is passed on in a family.

5
Association analysis, is population-based
Association analysis, is population-based. Association analysis does not depend explicitly on pedigrees but instead looks for increased or decreased frequency of a particular allele or set of alleles in a sample of affected individuals taken from the population, compared with a control set of unaffected people.

6
How Does Gene Mapping Contribute to Medical Genetics?
Disease gene mapping has immediate clinical application by providing information about a gene's location that can be used to develop indirect linkage methods for use in prenatal diagnosis, pre-symptomatic diagnosis, and carrier testing Disease gene mapping is a critical first step in identifying a disease gene. Mapping the gene focuses attention on a limited region of the genome in which to carry out a systematic analysis of all the genes so we can find the mutations or variants that contribute to the disease (known as positional cloning).
.")
7
Positional cloning of a disease gene provides an opportunity to characterize the disorder as to the extent of: locus heterogeneity, the spectrum of allelic heterogeneity, the frequency of various disease-causing or predisposing variants in various populations, the penetrance and positive predictive value of mutations, the fraction of the total genetic contribution to a disease attributable to the variant at any one locus, and the natural history of the disease in asymptomatic at-risk individuals.

8
Characterization of a gene and the mutations in it furthers our understanding of
disease pathogenesis development of specific and sensitive diagnosis by direct detection of mutations, population-based carrier screening to identify individuals at risk for disease in themselves or their offspring, development of cell and animal models, drug therapy to prevent or ameliorate disease or to slow its progression, and treatment by gene replacement

9
Independent Assortment and Homologous Recombination in Meiosis
The effect of recombination on the origin of various portions of a chromosome. Because of crossing over in meiosis, the copy of the chromosome the boy (generation III) inherited from his mother is a mosaic of segments of all four of his grandparents' copies of that chromosome.
 inherited from his mother is a mosaic of segments of all four of his grandparents copies of that chromosome.")
10
Since homologous chromosomes look identical under the microscope, we must be able to differentiate them in order to trace the grandparental origin of each segment, and to determine if and where recombination has occurred. Genetic marker: any characteristic located at the same position on a pair of homologous chromosomes and allows distinguishing them. Millions of genetic markers are now available that can be genotyped by PCR.

11
Alleles at Loci on Different Chromosomes Assort Independently
Independent assortment of alleles at two loci, 1 and 2, when they are located on different chromosomes. Assume that alleles D and M were inherited from one parent, d and m from the other Half (50%) of gametes will be parental (DM or dm) and half (50%) will be non-parental (dM or Dm).
 of gametes will be parental (DM or dm) and half (50%) will be non-parental (dM or Dm).")
12
Gametes containing DM or dm are non- recombinant
Assume D and M are paternally derived and d and m are maternally derived. Gametes containing DM or dm are non- recombinant Alleles at Loci on the Same Chromosome Assort Independently if at Least One Crossover Occurs Between Them in Every Meiosis Note: Genes that reside on the same chromosome are said to be syntenic

13
recombinant chromosome
If crossing over occurs at least once in the segment between the loci, the resulting chromatids may be either nonrecombinant or Dm and dM, which are not the same as the parental chromosomes; such a nonparental chromosome is therefore a recombinant chromosome

14
The ratio of recombinant to nonrecombinant genotypes will be, on average, 1 : 1, just as if the loci were on separate chromosomes and assorting independently

15
Recombination Frequency and Map Distance
Crossing over between homologous chromosomes in meiosis is shown in the quadrivalents on the left. Crossovers result in new combinations of maternally and paternally derived alleles on the recombinant chromosomes present in gametes. If no crossing over occurs in the interval between loci 1 and 2, only parental (nonrecombinant) allele combinations, DM and dm, occur in the offspring. If one or two crossovers occur in the interval between the loci, half the gametes will contain a nonrecombinant and half the recombinant combination. The same is true if more than two crossovers occur between the loci.
 allele combinations, DM and dm, occur in the offspring. If one or two crossovers occur in the interval between the loci, half the gametes will contain a nonrecombinant and half the recombinant combination. The same is true if more than two crossovers occur between the loci.")
16
The smaller the recombination frequency, the closer together two loci are.

17
A common notion for recombination frequency is θ, where θ varies from 0 (no recombination at all) to 0.5 (independent assortment).
 to 0.5 (independent assortment).")
18
Detecting the recombination events between loci requires that (1) a parent be heterozygous (informative) at both loci and (2) we know which allele at locus 1 is on the same chromosome as which allele at locus 2. Alleles on the same homologue are in coupling (or cis), whereas alleles on the different homologues are in repulsion (or trans).
, whereas alleles on the different homologues are in repulsion (or trans).")
19
Effect of Heterozygosity and Phase on Detecting Recombination Events
Possible phases of alleles M and m at a marker locus with alleles D and d at a disease locus

20
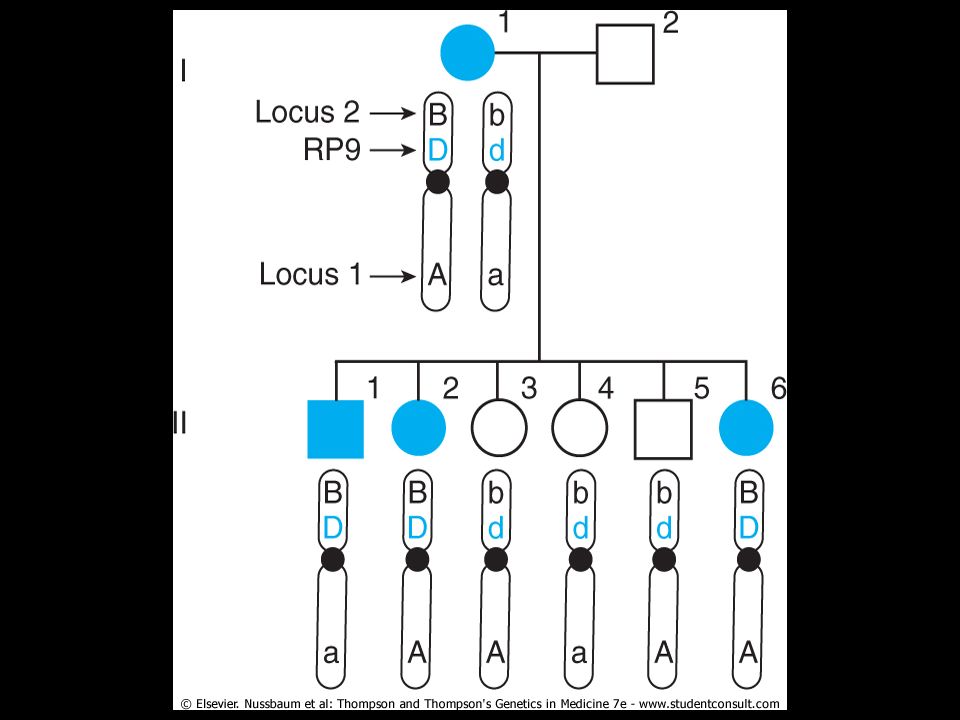
Co-inheritance of the gene for an autosomal dominant form of retinitis pigmentosa, RP9, with marker locus 2 and not with marker locus 1. Only the mother's contribution to the children's genotypes is shown. The mother (I-1) is affected with this dominant disease and is heterozygous at the RP9 locus (Dd) as well as at loci 1 and 2. She carries the A and B alleles on the same chromosome as the mutant RP9 allele (D). The unaffected father is homozygous normal (dd) at the RP9 locus as well as at the two marker loci (AA and BB); his contributions to his offspring are not considered further.
 is affected with this dominant disease and is heterozygous at the RP9 locus (Dd) as well as at loci 1 and 2. She carries the A and B alleles on the same chromosome as the mutant RP9 allele (D). The unaffected father is homozygous normal (dd) at the RP9 locus as well as at the two marker loci (AA and BB); his contributions to his offspring are not considered further.")
21
All three affected offspring have inherited the B allele at locus 2 from their mother, whereas the three unaffected offspring have inherited the b allele. Thus, all six offspring are nonrecombinant for RP9 and marker locus 2. However, individuals II-1, II-3, and II-5 are recombinant for RP9 and marker locus 1, indicating that meiotic crossover has occurred between these two loci.

22
Determine the phase in the II. 1
Determine the phase in the II.1. How many recombinants/Non-recombinants

23
Phase known/unknown

24
Detecting the recombination events between loci requires informative parent and knowing the phase

25
Linkage and Recombination Frequency
Linkage is the term used to describe a departure from the independent assortment of two loci, or, in other words, the tendency for alleles at loci that are close together on the same chromosome to be transmitted together, as an intact unit, through meiosis. Analysis of linkage depends on determining the frequency of recombination as a measure of how close different loci are to each other on a chromosome. If two loci are so close together that θ= 0 between them, they are said to be tightly linked; if they are so far apart that θ= 0.5, they are assorting independently and are unlinked.

26
Suppose that among the offspring of informative meioses (i. e
Suppose that among the offspring of informative meioses (i.e., those in which a parent is heterozygous at both loci), 80% of the offspring are non-recombinant and 20% are recombinant. At first glance, the recombination frequency is therefore 20% (θ= 0.2). the accuracy of this measure of depends on the size of the family used to make the measurement.
, 80% of the offspring are non-recombinant and 20% are recombinant. At first glance, the recombination frequency is therefore 20% (θ= 0.2). the accuracy of this measure of depends on the size of the family used to make the measurement.")
27
The map distance between two loci is a theoretical concept that is based on real data, the extent of observed recombination, θ, between the loci. Map distance is measured in units called centimorgans (cM), defined as the genetic length over which, on average, one crossover occurs in 1% of meioses. Therefore, a recombination fraction of 1% ( θ= 0.01) translates approximately into a map distance of 1 cM As the map distance between two loci increases, however, the frequency of recombination we observe between them does not increase proportionately (Fig. 10-7). This is because as the distance between two loci increases, the chance that the chromosome carrying these two markers could undergo more than one crossing over event between these loci also increases. As a rule of thumb, recombination frequency begins to underestimate true genetic distance significantly once rises above 0.1.
, defined as the genetic length over which, on average, one crossover occurs in 1% of meioses. Therefore, a recombination fraction of 1% ( θ= 0.01) translates approximately into a map distance of 1 cM. As the map distance between two loci increases, however, the frequency of recombination we observe between them does not increase proportionately (Fig. 10-7). This is because as the distance between two loci increases, the chance that the chromosome carrying these two markers could undergo more than one crossing over event between these loci also increases. As a rule of thumb, recombination frequency begins to underestimate true genetic distance significantly once rises above 0.1.")
28
The relationship between map distance in centimorgans and recombination fraction,θ. Recombination fraction (solid line) and map distance (dotted line) are nearly equal, with 1 cM = 0.01 recombination, for values of genetic distance below 10 cM, but they begin to diverge because of double crossovers as the distance between the markers increases. The recombination fraction approaches a maximum of 0.5 no matter how far apart loci are; the genetic distance increases proportionally to the distance between loci.
 and map distance (dotted line) are nearly equal, with 1 cM = 0.01 recombination, for values of genetic distance below 10 cM, but they begin to diverge because of double crossovers as the distance between the markers increases. The recombination fraction approaches a maximum of 0.5 no matter how far apart loci are; the genetic distance increases proportionally to the distance between loci..")
29
Genetic maps and physical maps
To measure true genetic map distance between two widely spaced loci accurately, therefore, one has to use markers spaced at short genetic distances in the interval between these two loci and add up the values of θ between the intervening markers. (Fig. 10-8). For example, human chromosome 1 is the largest human chromosome in physical length (283 Mb) and also has the greatest genetic length, 270 cM (0.95 cM/Mb); the q arm of the smallest chromosome, number 21, is 30 Mb in physical length and 62 cM in genetic length (∼2.1 cM/Mb). Overall, the human genome, which is estimated to contain about 3200 Mb, has a genetic length of 3615 cM, for an average of 1.13 cM/Mb. Furthermore, the ratio of genetic distance to physical length is not uniform along a chromosome as one looks with finer and finer resolution at recombination versus physical length.
. For example, human chromosome 1 is the largest human chromosome in physical length (283 Mb) and also has the greatest genetic length, 270 cM (0.95 cM/Mb); the q arm of the smallest chromosome, number 21, is 30 Mb in physical length and 62 cM in genetic length (∼2.1 cM/Mb). Overall, the human genome, which is estimated to contain about 3200 Mb, has a genetic length of 3615 cM, for an average of 1.13 cM/Mb. Furthermore, the ratio of genetic distance to physical length is not uniform along a chromosome as one looks with finer and finer resolution at recombination versus physical length.")
30
A diagram showing how adding together short genetic distances, measured as recombination fraction,θ , between neighboring loci A, B, C, and so on allows accurate determination of genetic distance between the two loci A and H located far apart. The value of between A and H is not an accurate measure of genetic distance.

31
Sex Differences in Map Distances
Just as male and female gametogenesis shows sex differences in the types of mutations and their frequencies, there are also significant differences in recombination between males and females. Across all chromosomes, the genetic length in females, 4460 cM, is 72% greater than the genetic distance of 2590 cM in males, and it is consistently about 70% greater in females on each of the different autosomes. The reason for increased recombination in females compared with males is unknown, although one might speculate that it has to do with the many years that female gamete precursors remain in meiosis I before ovulation.

32
Linkage Equilibrium and Disequilibrium
When a disease allele first enters the population (by mutation or a founder), the particular set of alleles at markers linked to the disease locus constitutes a disease-containing haplotype The degree to which this haplotype will persist as such over time depends on probability of recombination
, the particular set of alleles at markers linked to the disease locus constitutes a disease-containing haplotype. The degree to which this haplotype will persist as such over time depends on probability of recombination.")
33
The speed with which recombination will move disease allele onto a new haplotype is the product of two main factors: The number of generations, and therefore the number of opportunities for recombination The frequency of recombination between the loci A third factor, selection for or against a particular haplotype, but its effect has been difficult to prove in humans

35
The shorter the time since the disease allele appeared and the smaller the value of θ, the greater is the chance that the disease-containing haplotype will persist intact. With longer time periods and greater values of θ, shuffling will go to completion and the allele frequencies for marker alleles in the haplotype that includes the disease allele will come to equal the frequencies of these marker alleles in all chromosomes in the population i.e., alleles in the haplotype will have reached equilibrium.

36
The Haplotype Map (HapMap)
One of the biggest human genomics efforts to follow completion of the sequencing is a project designed to create a haplotype map (HapMap) of the genome. The goal of the HapMap project is to make LD measurements between a dense collection of millions of single nucleotide polymorphisms (SNPs) throughout the genome to delineate the genetic landscape of the genome on a fine scale. To accomplish this goal, geneticists collected and characterized millions of SNP loci, developed methods to genotype them rapidly and inexpensively, and used them, one pair at a time, to measure LD between neighboring markers throughout the genome. The measurements were made in samples that included both unrelated population samples and samples containing one child and both parents, obtained from four geographically distinct groups: a primarily European population, a West African population, a Han Chinese population, and a population from Japan
 of the genome. The goal of the HapMap project is to make LD measurements between a dense collection of millions of single nucleotide polymorphisms (SNPs) throughout the genome to delineate the genetic landscape of the genome on a fine scale. To accomplish this goal, geneticists collected and characterized millions of SNP loci, developed methods to genotype them rapidly and inexpensively, and used them, one pair at a time, to measure LD between neighboring markers throughout the genome. The measurements were made in samples that included both unrelated population samples and samples containing one child and both parents, obtained from four geographically distinct groups: a primarily European population, a West African population, a Han Chinese population, and a population from Japan.")
37
The study showed that: 1) More than 90% of all SNPs are shared among such geographocally disparate populations, with allele frequencies that are quite similar in the different populations This finding indicates that most SNPs are old and predate the waves of emigration out of East Africa that populated the rest of the world Differences in allele frequencies in a small fraction of SNPs may be the result of either genetic drift/founder effect or selection in localized geographical regions after migration out of Africa.
 More than 90% of all SNPs are shared among such geographocally disparate populations, with allele frequencies that are quite similar in the different populations. This finding indicates that most SNPs are old and predate the waves of emigration out of East Africa that populated the rest of the world. Differences in allele frequencies in a small fraction of SNPs may be the result of either genetic drift/founder effect or selection in localized geographical regions after migration out of Africa.")
38
Such SNPs, termed ancestry informative markers, are used in studies of human origin, migration and gene flow. In forensic investigations, to determine the likely ethnic background when the only available evidence is DNA

39
2) When pairwise measurements of LD were made for neighboring SNPs across the genome, contiguous SNPs can be grouped into clusters of varying size in which SNPs in any one cluster shows high levels of LD with each other. These clusters of SNPs in high LD, located across segments of a few kb to a few dozen Kb are termed LD blocks. The sizes of LD blocks are not identical in all populations. African populations have smaller blocks as compared to other populations.

40
A 145-kb region of chromosome 4 containing 14 SNPs
A 145-kb region of chromosome 4 containing 14 SNPs. In cluster 1, containing SNPs 1 through 9, five of the 29 = 512 theoretically possible haplotypes are responsible for 98% of all the haplotypes in the population, reflecting substantial linkage disequilibrium among these SNP loci. Similarly, in cluster 2, only three of the 24 = 16 theoretically possible haplotypes involving SNPs 11 to 14 represent 99% of all the haplotypes found. In contrast, alleles at SNP 10 are found in linkage equilibrium with the SNPs in cluster 1 and cluster 2.

41
3) Pairwise measurements of recombination between closely neighboring SNPs revealed that the ratio of map distance to base pairs was not constant (~1 cM/Mb). Instead ranged from far below 0.01 cM/Mb to more than 60 cM/Mb. - This indicates that rate of recombination between polymorphic markers which was thought to be uniform is, in fact, the result of an averaging of “hotspots” of recombination interspersed among regions of little or no recombination.

42
B, A schematic diagram in which each box contains the pairwise measurement of the degree of linkage disequilibrium between two SNPs (e.g., the arrow points to the box, outlined in black, containing the value of D' for SNPs 2 and 7). The higher the degree of LD, the darker the color in the box, with maximum D' values of 1.0 occurring when there is complete LD. Two LD blocks are detectable, the first containing SNPs 1 through 9, and the second SNPs 11 through 14. In the first block, pairwise measurements of D' reveal LD. A similar level of LD is found in block 2. Between blocks, the 14-kb region containing SNP 10 shows no LD with neighboring SNPs 9 or 11 or with any of the other SNP loci. Below is a graph of the ratio of map distance to physical distance (cM/Mb) showing that a recombination hotspot is present in the region around SNP 10 between the two blocks, with values of recombination that are 50- to 60-fold above the average of approximately 1.13 cM/Mb for the genome.
 showing that a recombination hotspot is present in the region around SNP 10 between the two blocks, with values of recombination that are 50- to 60-fold above the average of approximately 1.13 cM/Mb for the genome..")
43
MAPPING HUMAN GENES BY LINKAGE ANALYSIS
Determining Whether Two Loci Are Linked Linkage analysis is a method of mapping genes that uses family studies to determine whether two genes show linkage (are linked) when passed on from one generation to the next. To decide whether two loci are linked, and if so, how close or far apart they are, we rely on two pieces of information. First, we ascertain whether the recombination fraction between two loci deviates significantly from 0.5 Second, if θ is less than 0.5, we need to make the best estimate we can of θ since that will tell us how close or far apart the linked loci are.
 when passed on from one generation to the next. To decide whether two loci are linked, and if so, how close or far apart they are, we rely on two pieces of information. First, we ascertain whether the recombination fraction between two loci deviates significantly from 0.5. Second, if θ is less than 0.5, we need to make the best estimate we can of θ since that will tell us how close or far apart the linked loci are.")
44
Likelihoods are probability values; odds are ratios of likelihoods.
For these determinations, a statistical tool called the likelihood ratio is used. Likelihoods are probability values; odds are ratios of likelihoods. One proceeds as follows: examine a set of actual family data, count the number of children who show or do not show recombination between the loci, calculate the likelihood of observing the data at various possible values of θ between 0 and 0.5. Calculate a second likelihood based on the null hypothesis that the two loci are unlinked, that is, θ= 0.50. take the ratio of the likelihood of observing the family data for various values of θ to the likelihood the loci are unlinked to create an odds ratio.

45
The computed odds ratios for different values of are usually expressed as the log10 of this ratio and are called a LOD score (Z) for "logarithm of the odds."
 for logarithm of the odds.")
46
Model-Based Linkage Analysis of Mendelian Diseases
Linkage analysis is called model-based (or parametric) when it assumes that there is a particular mode of inheritance (autosomal dominant, autosomal recessive, or X-linked) that explains the inheritance pattern. LOD score analysis allows mapping of genes in which mutations cause diseases that follow mendelian inheritance. The LOD score gives both: a best estimate of the recombination frequency, θmax, between a marker locus and the disease locus; and
 when it assumes that there is a particular mode of inheritance (autosomal dominant, autosomal recessive, or X-linked) that explains the inheritance pattern. LOD score analysis allows mapping of genes in which mutations cause diseases that follow mendelian inheritance. The LOD score gives both: a best estimate of the recombination frequency, θmax, between a marker locus and the disease locus; and.")
47
an assessment of how strong the evidence is for linkage at that value of θmax. Values of the LOD score above 3 are considered strong evidence. Linkage at a particular θ max of a disease gene locus to a marker with known physical location implies that the disease gene locus must be near the marker

48
The odds ratio is important in two ways.
First, it provides a statistically valid method for using the family data to estimate the recombination frequency between the loci. If θ max differs from 0.50, you have evidence of linkage. However, even if θ max is the best estimate you can make, how good an estimate is it? The odds ratio also provides you with an answer to this question because the higher the value of Z, the better an estimate θmax is. Positive values of Z (odds >1) at a given θ suggest that the two loci are linked, whereas negative values (odds <1) suggest that linkage is less likely than the possibility that the two loci are unlinked. By convention, a combined LOD score of +3 or greater (equivalent to greater than 1000:1 odds in favor of linkage) is considered definitive evidence that two loci are linked.
 at a given θ suggest that the two loci are linked, whereas negative values (odds <1) suggest that linkage is less likely than the possibility that the two loci are unlinked. By convention, a combined LOD score of +3 or greater (equivalent to greater than 1000:1 odds in favor of linkage) is considered definitive evidence that two loci are linked.")
49
Mapping genes by linkage analysis provides an opportunity to localize medically relevant genes by following inheritance of the condition and the inheritance of alleles at polymorphic markers to see if the disease locus and the polymorphic marker locus are linked. Return to the family shown in Figure The mother has an autosomal dominant form of retinitis pigmentosa. She is also heterozygous for two loci on chromosome 7, one at 7p14 and one at the distal end of the long arm. One can see that transmission of the RP mutant allele (D) invariably "follows" that of allele B at marker locus 2 from the first generation to the second generation in this family. All three offspring with the disease (who therefore must have inherited their mother's mutant allele D at the RP locus) also inherited the B allele at marker locus 2. All the offspring who inherited their mother's normal allele, d, inherited the b allele and will not develop RP. The gene encoding RP, however, shows no tendency to follow the allele at marker locus 1.
 invariably follows that of allele B at marker locus 2 from the first generation to the second generation in this family. All three offspring with the disease (who therefore must have inherited their mother s mutant allele D at the RP locus) also inherited the B allele at marker locus 2. All the offspring who inherited their mother s normal allele, d, inherited the b allele and will not develop RP. The gene encoding RP, however, shows no tendency to follow the allele at marker locus 1.")
51
Suppose we let θ be the "true" recombination fraction between RP and locus 2, the fraction we would see if we had unlimited numbers of offspring to test. Because either a recombination occurs or it does not, the probability of a recombination, θ, and the probability of no recombination must add up to 1. Therefore, the probability that no recombination will occur is 1 -θ . There are only six offspring, all of whom show no recombination. Because each meiosis is an independent event, one multiplies the probability of a recombination, θ, or of no recombination, (1 -θ ), for each child. The likelihood of seeing zero offspring with a recombination and six offspring with no recombination between RP and marker locus 2 is therefore (θ)0(1 -θ )6. The LOD score between RP and marker 2, then, is: The maximum value of Z is 1.81, which occurs when = 0, and is suggestive of but not definite evidence for linkage because Z is positive but less than 3.
, for each child. The likelihood of seeing zero offspring with a recombination and six offspring with no recombination between RP and marker locus 2 is therefore (θ)0(1 -θ )6. The LOD score between RP and marker 2, then, is: The maximum value of Z is 1.81, which occurs when = 0, and is suggestive of but not definite evidence for linkage because Z is positive but less than 3.")
52
Combining LOD Score Information Across Families
In the same way that each meiosis in a family that produces a nonrecombinant or recombinant offspring is an independent event, so too are the meioses that occur in other families. We can therefore multiply the likelihoods in the numerators and denominators of each family's likelihood odds ratio together. An equivalent but more convenient calculation is to add the log10 of each likelihood ratio, calculated at the various values of θ, to form an overall Z score for all families combined.

53
In the case of RP in Figure 10-6, suppose two other families were studied and one showed no recombination between locus 2 and RP in four children and the third showed no recombination in five children. The individual LOD scores can be generated for each family and added together (Table 10-1). In this case, one could say that the RP gene in this group of families is linked to locus 2. Because the chromosomal location of polymorphic locus 2 was known to be at 7p14, the RP in this family can be mapped to the region around 7p14, which is near RP9, an already identified locus for one form of autosomal dominant RP.
. In this case, one could say that the RP gene in this group of families is linked to locus 2. Because the chromosomal location of polymorphic locus 2 was known to be at 7p14, the RP in this family can be mapped to the region around 7p14, which is near RP9, an already identified locus for one form of autosomal dominant RP..")
54
Table 10-1. LOD Score Table for Three Families with Retinitis Pigmentosa
θ=0.4 θ=0.3 θ=0.2 θ=0.1 θ=0.05 θ=0.01 θ=0 0.48 0.88 1.22 1.53 1.67 1.78 1.8 Family 1 0.32 0.58 0.82 1.02 1.11 1.19 1.2 Family 2 0.39 0.73 1.28 1.39 1.48 1.5 Family 3 2.19 3.06 2.83 4.17 4.45 4.5 Total Zmax = 4.5 at θ max = 0

55
If, however, some of the families being used for the study have RP due to mutations at another locus, the LOD scores between families will diverge, with some showing a trend to being positive at small values of θ and others showing strongly negative LOD scores at these values. One can still add the Z scores together, but the result will show a sharp decline in the overall LOD score. Thus, in linkage analysis involving more than one family, unsuspected locus heterogeneity can obscure what may be real evidence for linkage in a subset of families.

56
Phase information is important in linkage analysis
Phase information is important in linkage analysis. Figure shows two pedigrees of autosomal dominant neurofibromatosis, type 1 (NF1). In the three-generation family on the left, the affected mother, II-2, is heterozygous at both the NF1 locus (D/d) and a marker locus (M/m), but we have no genotype information on her parents. Her unaffected husband, II-1, is homozygous both for the normal allele d at the NF1 locus and happens to be homozygous for allele M at the marker locus. He can only transmit to his offspring a chromosome that has the normal allele (d) and the M allele.
. In the three-generation family on the left, the affected mother, II-2, is heterozygous at both the NF1 locus (D/d) and a marker locus (M/m), but we have no genotype information on her parents. Her unaffected husband, II-1, is homozygous both for the normal allele d at the NF1 locus and happens to be homozygous for allele M at the marker locus. He can only transmit to his offspring a chromosome that has the normal allele (d) and the M allele.")
57
By inspection, then, we can infer which alleles in each child have come from the mother. The two affected children received the m alleles along with the D disease allele, and the one unaffected has received the M allele along with the normal d allele. Without knowing the phase of these alleles in the mother, either all three offspring are recombinants or all three are nonrecombinants.

58
Figure Two pedigrees of autosomal dominant neurofibromatosis, type 1 (NF1). A, Phase of the disease allele D and marker alleles M and m in individual II-2 is unknown. B, Availability of genotype information for generation I allows a determination that the disease allele D and marker allele M are in coupling in individual II-2. NR, nonrecombinant; R, recombinant.

59
Which of these two possibilities is correct
Which of these two possibilities is correct? There is no way to know for certain, and thus we must compare the likelihoods of the two possible results. Given that II-2 is an M/m heterozygote, we assume the correct phase on her two chromosomes is D-m and d-M half of the time and D-M and d-m the other half. If the phase of the disease allele is D-m, all three children have inherited a chromosome in which no recombination occurred between NF1 and the marker locus. If the probability of recombination between NF1 and the marker is θ, the probability of no recombination is (1 - θ), and the likelihood of having zero recombinant and three nonrecombinant chromosomes is θ0 (1-θ)3.
, and the likelihood of having zero recombinant and three nonrecombinant chromosomes is θ0 (1-θ)3.")
60
The contribution to the total likelihood, assuming this phase is correct half the time, is 1/2 θ0 (1-θ)3.The other half of the time, however, the correct phase is D-M and d-m, which makes each of these three children recombinants; the likelihood, assuming this phase is correct half the time, is 1/2 θ3(1-θ)0. To calculate the overall likelihood of this pedigree, we add the likelihood calculated assuming one phase in the mother is correct to the likelihood calculated assuming the other phase is correct. Therefore, the overall likelihood = 1/2(1-θ)3 + 1/2 (θ3)/1/8
3 + 1/2 (θ3)/1/8.")
61
By evaluating the relative odds for values of θ from 0 to 0
By evaluating the relative odds for values of θ from 0 to 0.5, the maximum value of the LOD score, Zmax, is found to be log(4) = (when θ = 0.0) Table Because this is far short of a LOD score greater than 3, we would need at least five equivalent families to establish linkage (at θ = 0.0) between this marker locus and NF1. With slightly more complex calculations (made much easier by computer programs written to facilitate linkage analysis), one can calculate the LOD scores for other values of θ (see Table 10-2).
 = (when θ = 0.0) Table Because this is far short of a LOD score greater than 3, we would need at least five equivalent families to establish linkage (at θ = 0.0) between this marker locus and NF1. With slightly more complex calculations (made much easier by computer programs written to facilitate linkage analysis), one can calculate the LOD scores for other values of θ (see Table 10-2).")
62
Why are the two phases in individual II-2 in the pedigree shown in Figure 10-14A equally likely?
First, unless the marker locus and NF1 are so close together as to produce linkage disequilibrium between alleles at these loci, we would expect them to be in linkage equilibrium. Second, new mutations represent a substantial fraction of all the alleles in an autosomal dominant disease with reduced fitness, such as NF1. If new mutations are occurring independently and repeatedly, the alleles that happened to be present at the neighboring linked loci when each mutation occurred in the NF1 gene will then be the alleles in coupling with the new disease mutation. A group of unrelated families are likely to have many different mutant alleles, each of which is as likely to be in coupling with one polymorphic marker allele at a linked locus as with any other.

63
Suppose now that additional genotype information, shown in Figure 10-14B, becomes available in the family in Figure A. By inspection, it is now clear that the maternal grandfather, I-1, must have transmitted both the NF1 allele (D) and the M allele to his daughter. This finding does not require any assumption about whether a crossover occurred in the grandfather's germline; all that matters is that we can be sure the paternally derived chromosome in individual II-2 must have been D-M and the maternally derived chromosome was d-m.

64
The availability of genotypes in the first generation makes this a phase-known pedigree.
The three children can now be scored definitively as nonrecombinant and we do not have to consider the opposite phase. The probability of having three children with the observed genotypes is now (1 -θ )3. As in the previous phase-unknown pedigree, the probability of the observed data if there is no linkage between the loci is (1/2)3 = 1/8. Overall, the relative odds for this pedigree are (1 - θ)3 ÷ 1/8 in favor of linkage, and the maximum LOD score Z at θ= 0.0 is or 8 to 1 (see Table 10-2). Thus, the strength of the evidence supporting linkage (8 to 1) is twice as great in the phase- known situation as in the phase-unknown situation (4 to 1).
3. As in the previous phase-unknown pedigree, the probability of the observed data if there is no linkage between the loci is (1/2)3 = 1/8. Overall, the relative odds for this pedigree are (1 - θ)3 ÷ 1/8 in favor of linkage, and the maximum LOD score Z at θ= 0.0 is or 8 to 1 (see Table 10-2). Thus, the strength of the evidence supporting linkage (8 to 1) is twice as great in the phase- known situation as in the phase-unknown situation (4 to 1).")
65
Determining Phase from Pedigrees
As shown in the pedigree in Figure 10-14B, having grandparental genotypes may be helpful in establishing phase in the next generation. However, depending on what the genotypes are, phase may not always be definitively determined. For example, if the grandmother, I-2, had been an M/m heterozygote, it would not be possible to determine the phase in the affected parent, individual II-2.

66
Determining Phase from Pedigrees
For linkage analysis in X-linked pedigrees, the mother's father's genotype is particularly important because, as illustrated in Figure 10-15, it provides direct information on linkage phase in the mother. Because there can be no recombination between X- linked genes in a male and because the mother always receives her father's only X, any X-linked marker present in her genotype, but not in her father's, must have been inherited from her mother. Knowledge of phase, so important for genetic counseling, can thus be readily ascertained from the appropriate male members of an X-linked pedigree, if they are available for study.

67
Pedigree of X-linked hemophilia.
The affected grandfather in the first generation has the disease (mutant allele h) and is hemizygous for allele M at an X-linked locus. No matter how far apart the marker locus and the factor VIII gene are on the X, there is no recombination involving the X-linked portion of the X chromosome in a male, and he will pass the hemophilia mutation h and allele M together. The phase in his daughter must be that h and M are in coupling
 and is hemizygous for allele M at an X-linked locus. No matter how far apart the marker locus and the factor VIII gene are on the X, there is no recombination involving the X-linked portion of the X chromosome in a male, and he will pass the hemophilia mutation h and allele M together. The phase in his daughter must be that h and M are in coupling.")
68
MAPPING OF COMPLEX TRAITS
Two major approaches to locate and identify genes that predispose to complex disease or contribute to genetic variance of quantitative traits Affected pedigree member method: if a region of genome is shared more frequently than expected by relatives concordant for a particular disease, the inference is alleles predispose to disease at one or more loci in that region. Association: looks for increased frequency of particular alleles in affected compared with unaffected in the pop.

69
Model-Free Linkage Analysis of Complex Traits
Linkage analysis is called model-free (or nonparametric) when it does not assume any particular mode of inheritance to explain the inheritance pattern. Nonparametric LOD (NPL) score analysis allows mapping of genes in which variants contribute to susceptibility for diseases (so-called qualitative traits) or to physiological measurements (known as quantitative traits) that do not follow a straightforward mendelian inheritance pattern.
 when it does not assume any particular mode of inheritance to explain the inheritance pattern. Nonparametric LOD (NPL) score analysis allows mapping of genes in which variants contribute to susceptibility for diseases (so-called qualitative traits) or to physiological measurements (known as quantitative traits) that do not follow a straightforward mendelian inheritance pattern.")
70
MAPPING OF COMPLEX TRAITS
NPL scores are based on testing for excessive allele- sharing among relatives, such as pairs of siblings, who are both affected with a disease or who show greater similarity to each other for some quantitative trait compared with the average for the population.

71
MAPPING OF COMPLEX TRAITS
The NPL score gives an assessment of how strong the evidence is for increased allele sharing near polymorphic markers. A value of the NPL score greater than 3.6 is considered evidence for increased allele-sharing; an NPL score greater than 5.4 is considered strong evidence.

72
Model-Free Linkage Analysis of Qualitative (Disease) Traits
One type of model-free analysis is the affected sibpair method. Only siblings concordant for a disease are used No assumptions need be made about the number of loci involved or the inheritance pattern. Sibs are analyzed to determine whether there are loci at which affected sibpairs share alleles more frequently than expected by chance alone In this method, DNA of affected sibs is systematically analyzed by use of hundreds of polymorphic markers throughout the entire genome in a search for regions that are shared by the two sibs significantly more frequently than is expected on a purely random basis.

73
When elevated degrees of allele-sharing are found at a polymorphic marker, it suggests that a locus involved in the disease is located close to the marker. Whether the degree of allele-sharing diverges significantly from the expected by chance alone can be assessed by use of a maximum likelihood odds ratio to generate a nonparametric LOD score for excessive allele sharing.

74
Model-Free Linkage Analysis of Quantitative Traits
Model-free linkage methods based on allele-sharing can also be used to map loci involved in quantitative complex traits. Although a number of approaches are available, one interesting example is the highly discordant sibpair method. Once again, no assumptions need be made about the number of loci involved or the inheritance pattern. Sibpairs with values of a physiological measurement that are at opposite ends of the bell-shaped curve are considered discordant for that quantitative trait and can be assumed to be less likely to share alleles at loci that contribute to the trait. The DNA of highly discordant sibs is then systematically analyzed by use of polymorphic markers throughout the entire genome in a search for regions that are shared by the two sibs significantly less frequently than is expected on a purely random basis. When reduced levels of allele-sharing are found at a polymorphic marker, it suggests that the marker is linked to a locus whose alleles contribute to whatever physiological measurement is under study.

75
Disease Association An entirely different approach to identification of the genetic contribution to complex disease relies on finding particular alleles that are associated with the disease. The presence of a particular allele at a locus at increased or decreased frequency in affected individuals compared with controls is known as a disease association. In an association study, the frequency of a particular allele (such as for an HLA haplotype or a particular SNP or SNP haplotype) is compared among affected and unaffected individuals in the population
 is compared among affected and unaffected individuals in the population.")
76
Disease Association total control patients a+b b a With allele c+d d c
Without allele b+d a+c The RRR is approximately equal to the odds ratio when the disease is rare (i.e., a < b and c < d). (Do not confuse RRR (relative risk ratio) with λr, the risk ratio in relatives. λr is the prevalence of a particular disease phenotype in an affected individual's relatives versus that in the general population.)
. (Do not confuse RRR (relative risk ratio) with λr, the risk ratio in relatives. λr is the prevalence of a particular disease phenotype in an affected individual s relatives versus that in the general population.)")
77
If the study design is a case-control study in which individuals with the disease are selected in the population, a matching group of controls without disease are then chosen, and the genotypes of individuals in the two groups are determined; an association between disease and genotype is then calculated by an odds ratio. Odds are ratios. With use of the above table, the odds of an allele carrier's developing the disease is the number of allele carriers that develop the disease (a) divided by the number of allele carriers who do not develop the disease (b). Similarly, the odds of a noncarrier's developing the disease is the number of noncarriers who develop the disease (c) divided by the number of noncarriers who do not develop the disease (d). The disease odds ratio is then the ratio of these odds, that is, a ratio of ratios.
 divided by the number of allele carriers who do not develop the disease (b). Similarly, the odds of a noncarrier s developing the disease is the number of noncarriers who develop the disease (c) divided by the number of noncarriers who do not develop the disease (d). The disease odds ratio is then the ratio of these odds, that is, a ratio of ratios.")
78
If the study was designed as a cross-sectional or cohort study, in which a random sample of the entire population is chosen and then analyzed both for disease and for the presence of the susceptibility genotype, the strength of an association can be measured by the relative risk ratio (RRR). The RRR compares the frequency of disease in all those who carry a susceptibility allele ([a/(a + b)] with the frequency of disease in all those who do not carry a susceptibility allele ([c/(c + d)].
] with the frequency of disease in all those who do not carry a susceptibility allele ([c/(c + d)].")
79
The RRR is approximately equal to the odds ratio when the disease is rare (i.e., a < b and c < d). The significance of any association can be assessed in one of two ways: One is simply to ask if the values of a, b, c, and d differ from what would be expected if there were no association by a χ2 test. The other is determined by a 95% confidence interval for the relative risk ratio. This interval is the range in which one would expect the RRR to fall 95% of the time that you genotype a similar group of cases and controls by chance alone. If the frequency of the allele in question were the same in patients and controls, the RRR would be 1. Therefore, when the 95% confidence interval excludes the value of 1, then the RRR deviates from what would be expected for no association with P value <0.05.

80
, Example suppose there were a case-control study in which a group of 120 patients with cerebral vein thrombosis(CVT) Total Controls without CVT Patients with CVT 27 4 23 20210G > A allele present 213 116 97 20210G > A allele absent 240 120

81
For example, suppose there were a case-control study in which a group of 120 patients with cerebral vein thrombosis (CVT) and 120 matched controls were genotyped for the 20210G > A allele in the prothrombin gene. There is clearly a significant increase in the number of patients carrying the G > A allele versus controls (χ2 = 15 with 1 df; P < 10-10). Since this is a case-control study, we use an odds ratio (OR) to assess the strength of the association.
. Since this is a case-control study, we use an odds ratio (OR) to assess the strength of the association.")
82
Genome-Wide Association and the Haplotype Map
Association studies for human disease genes have been limited to particular sets of variants in restricted sets of genes. For example, geneticists might look for association with variants in genes encoding proteins thought to be involved in a pathophysiological pathway in a disease. Many such association studies were undertaken before the Human Genome Project era, with use of the HLA loci, because these loci are highly polymorphic and easily genotyped in case-control studies.

83
Genome-Wide Association and the Haplotype Map
A more powerful approach, however, would be to test systematically for association genome-wide between the more than 10 million variants in the genome and a disease phenotype, without any preconception of what genes and genetic variants might be contributing to the disease.

84
Tag SNPs By examining all the haplotypes within an LD block and measuring the degree of LD between them, it is possible to identify the most useful, minimum set of SNP alleles (so-called tag SNPs) that are capable of defining most of the haplotypes contained in each LD with minimum redundancy. In theory, a set of well-chosen tag-SNPs constitutes the minimal numbers of SNPs that need to be genotyped to provide nearly complete info on which haplotypes are present on any chromosome
 that are capable of defining most of the haplotypes contained in each LD with minimum redundancy. In theory, a set of well-chosen tag-SNPs constitutes the minimal numbers of SNPs that need to be genotyped to provide nearly complete info on which haplotypes are present on any chromosome.")
85
In practice, genotyping a few hundred thousand tag SNPs is only a bit less useful for an association study than is genotyping more than 10 million SNP genotypes at every known variant in the genome. Tag SNPs need to be examined and refined before we know if the results based on the four populations studied in the Hap-Map project are applicable world-wide.

86
From Gene Mapping to Gene Identification
Positional cloning: Mapping location of a disease gene by linkage analysis or other means, followed by identifying the gene on basis of its map position This strategy has led to identification of genes associated with hundreds of mendelian disorders and to a small but increasing number of genes associated with complex disorders.

87
Autozygosity mapping The method of choice for the discovery of autosomal recessive gene loci. Autozygosity is a term used to describe homozygosity for markers identical by descent (IBD) i.e., the two alleles are both copies of one specific allele that was present in a recent ancestor. An individual who is homozygous for two such alleles is said to be autozygous at that particular locus.
 i.e., the two alleles are both copies of one specific allele that was present in a recent ancestor. An individual who is homozygous for two such alleles is said to be autozygous at that particular locus.")
88
Autozygosity mapping

89
The methodology seeks homozygous regions in consanguineous families.
The rarer the alleles are in a population, the more likely the homozygosity represents autozygosity. The greater the number of affected individuals who have a shared homozygous region and the greater the size of the region, the more likely it is to harbor the mutation that causes the disease. DNA from the affected individuals is genotyped using SNP arrays/STRs and regions of homozygosity highlighted using IBD finder software. Linkage is confirmed using polymorphic more microsatellite markers Candidate genes within these haplotype regions are screened for pathogenic mutations.

Similar presentations



 — ————— Two homologous chromosomes,>")


BB. Terminology Genes Chromosomes Autosome Sex chromosome Locus Alleles Homozygous Heterozygous.>")













