Download presentation
Presentation is loading. Please wait.

1
Protein structure prediction.

2
Protein domains can be defined based on: Geometry: group of residues with the high contact density, number of contacts within domains is higher than the number of contacts between domains. - chain continuous domains - chain discontinous domains Kinetics: domain as an independently folding unit. Physics: domain as a rigid body linked to other domains by flexible linkers. Genetics: minimal fragment of gene that is capable of performing a specific function.

3
Domains as recurrent units of proteins. The same or similar domains are found in different proteins. Each domain has a well determined compact structure and performs a specific function. Proteins evolve through the duplication and domain shuffling. Protein domain classification based on comparing their recurrent sequence, structure and functional features – Conserved Domain Database

4
Protein folds. Fold definition: two folds are similar if they have a similar arrangement of SSEs (architecture) and connectivity (topology). Sometimes a few SSEs may be missing. Fold classification: structural similarity between folds is searched using structure- structure comparison algorithms.
 and connectivity (topology). Sometimes a few SSEs may be missing. Fold classification: structural similarity between folds is searched using structure- structure comparison algorithms..")
5
Definition of protein folds. Protein fold – arrangement of secondary structures into a unique topology/tertiary structure. Example of alpha+beta proteins: TIM beta/alpha-barrel contains parallel beta-sheet barrel, closed; n=8, S=8;TIM beta/alpha-barrel strand order 12345678, surrounded by alpha-helices NAD(P)-binding Rossmann-fold domains core: 3 layers, a/b/a; parallel beta-sheet of 6 strands,NAD(P)-binding Rossmann-fold domains order 321456
-binding Rossmann-fold domains core: 3 layers, a/b/a; parallel beta-sheet of 6 strands,NAD(P)-binding Rossmann-fold domains order")
6
Fold recognition. Unsolved problem: direct prediction of protein structure from the physico-chemical principles. Solved problem: to recognize, which of known folds are similar to the fold of unknown protein. Fold recognition is based on observations/assumptions: -The overall number of different protein folds is limited (1000-3000 folds) -The native protein structure is in its ground state (minimum energy)
 -The native protein structure is in its ground state (minimum energy).")
7
Protein structure prediction flowchart Protein sequence Database similarity search Does sequence align with a protein of known structure ? Protein family analysis Relationship to known structure? Three-dimensional comparative modeling Predicted three- dimensional structural model Structural analysis Is there a predicted structure? Three- dimensional structural analysis in laboratory No Yes NoYes No From D.W.Mount

8
Protein structure prediction. Prediction of three-dimensional structure from its protein sequence. Different approaches: -Homology modeling (predicted structure has a very close homolog in the structure database). -Fold recognition (predicted structure has an existing fold). -Ab initio prediction (predicted structure has a new fold).
. -Fold recognition (predicted structure has an existing fold). -Ab initio prediction (predicted structure has a new fold)..")
9
Homology modeling. Aims to produce protein models with accuracy close to experimental and is used for: -Protein structure prediction -Drug design -Prediction of functionally important sites (active or binding sites)
.")
10
Steps of homology modeling. 1.Template recognition & initial alignment. 2.Backbone generation. 3.Loop modeling. 4.Side-chain modeling. 5.Model optimization.

11
1. Template recognition. Recognition of similarity between the target and template. Target – protein with unknown structure. Template – protein with known structure. Main difficulty – deciding which template to pick, multiple choices/template structures. Template structure can be found by searching for structures in PDB using sequence-sequence alignment methods.

12
Two zones of sequence alignment. Two sequences are guaranteed to fold into the same structure if their length and sequence identity fall into “safe” zone. 50100150200 50 100 Homology modeling zone Twilight zone Alignment length Sequence identity

13
2. Backbone generation. If alignment between target and template is ready, copy the backbone coordinates of those template residues that are aligned. If two aligned residues are the same, copy their side chain coordinates as well.

14
3. Insertions and deletions. insertion AHYATPTTT AH---TPSS deletion Occur mostly between secondary structures, in the loop regions. Loop conformations – difficult to predict. Approaches to loop modeling: -Knowledge-based: searches the PDB for loops with known structure -Energy-based: an energy function is used to evaluate the quality of a loop. Energy minimization or Monte Carlo.

15
4. Side chain modeling. Side chain conformations – rotamers. In similar proteins - side chains have similar conformations. If % identity is high - side chain conformations can be copied from template to target. If % identity is not very high - modeling of side chains using libraries of rotamers and different rotamers are scored with energy functions. Problem: side chain configurations depend on backbone conformation which is predicted, not real E1E1 E2E2 E3E3 E = min(E1, E2, E3)
.")
16
5. Model optimization. Energy optimization of entire structure. Since conformation of backbone depends on conformations of side chains and vice versa - iteration approach: Predict rotamersShift in backbone

17
Classwork I: Homology modeling. -Go to NCBI Entrez, search for gi461699 -Do Blast search against PDB -Repeat the same for gi60494508 -Compare the results

18
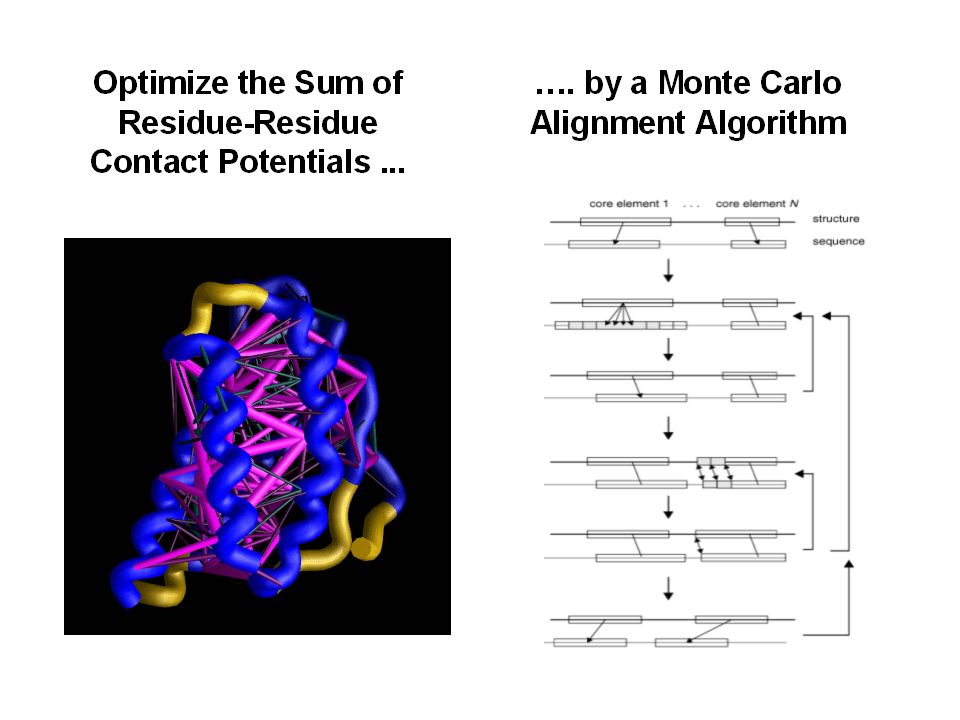
Fold recognition. Goal: to find protein with known structure which best matches a given sequence. Since similarity between target and the closest to it template is not high, sequence-sequence alignment methods fail. Solution: threading – sequence-structure alignment method.

19
Threading – method for structure prediction. Sequence-structure alignment, target sequence is compared to all structural templates from the database. Requires: -Alignment method (dynamic programming, Monte Carlo,…) -Scoring function, which yields relative score for each alternative alignment
 -Scoring function, which yields relative score for each alternative alignment.")
20
Scoring function for threading. Contact-based scoring function depends on the amino acid types of two residues and distance between them. Sequence-sequence alignment scoring function does not depend on the distance between two residues. If distance between two non- adjacent residues in the template is less than 8 Å, these residues make a contact.

21
Scoring function for threading. Ala Ile Tyr Trp w is calculated from the frequency of amino acid contacts in PDB; a i – amino acid type of target sequence aligned with the position “i” of the template; N- number of contacts

22
Classwork I: calculate the score for target sequence “ATPIIGGLPY” aligned to template structure which is defined by the contact matrix. 12345678910 1*** 2 3* 4* 5** 6* 7* 8* 9 ** ATPYIGL A-0.2-0.10 0.5-0.20.2 T0.3-0.1-0.2-0.30.10 P-0.2-0.4-0.10.1-0.2 Y-0.4-0.2-0.1-0.2 I0.30.20.4 G 0.2 L0.3

23
Alignment algorithms. Dynamic programming. “frozen approximation”: traceback in the alignment matrix is not possible for interactions between two amino acids, so that: b – amino acid type from template, not from target; now the score of every position does not depend on the alignment elsewhere in the sequence. Monte Carlo

25
CASP prediction competitions.

26
Threading model validation. Correct bond length and bond angles Correct placement of functionally important sites Prediction of global topology, not partial alignment (minimum number of gaps) >> 3.8 Angstroms
 >> 3.8 Angstroms.")
27
Placement of functionally important sites in threading. Prediction of structure of methylglyoxal synthase based on the template of carabamoyl phosphate synthase

28
Classwork II: Homology modeling. -Go to NCBI Entrez, search for gi461699 -Do Blast search against PDB -Repeat the same for gi60494508 -Predict functionally important sites

29
GenThreader http://bioinf.cs.ucl.ac.uk/psipred. http://bioinf.cs.ucl.ac.uk/psipred 1.Predicts secondary structures for target sequence. 2.Makes sequence profiles (PSSMs) for each template sequence. 3.Uses threading scoring function to find the best matching profile.
 for each template sequence. 3.Uses threading scoring function to find the best matching profile..")
30
Classwork III. -Go to http://bioinf.cs.ucl.ac.uk/psipredhttp://bioinf.cs.ucl.ac.uk/psipred -Go over the options of protein structure prediction program -Predict structure for protein sequence (“gwu_thread_seq.txt”) http://bioinf2.cs.ucl.ac.uk/psiout/29594540 ad0cf784.gen.html
 ad0cf784.gen.html.")
31
Protein engineering and protein design. Protein engineering – altering protein sequence to change protein function or structure Protein design – designing de novo protein which satisfies a given requirement

32
Protein engineering strategies. Goals: Design proteins with certain function Increase activity of enzymes Increase binding affinity and specificity of proteins Increase protein stability Design proteins which bind novel ligands

33
Protein engineering uses combinatorial libraries. Random mutagenesis introduces different mutations in many genes of interest. Active proteins are separated from inactive ones: - in vivo (measuring effect on the whole cell) - in vitro (phage display, gene is inserted into phage DNA, expressed, selected if it binds immobilized target protein)
 - in vitro (phage display, gene is inserted into phage DNA, expressed, selected if it binds immobilized target protein).")
34
Specificity of Kunitz inhibitors can be optimized by protein engineering. Kunitz domains – specific inhibitors of trypsin-like proteinases, highly conserved structure with only 33% identity. Each Kunitz domain recognizes one or more proteinases through the binding loop (yellow). Phage display method found mutants of Kunitz inhibitors which have higher specificity than native ones. Modeling of mutant proteins showed that enhanced specificity is caused by increased complementarity between binding loop and the active site.
. Phage display method found mutants of Kunitz inhibitors which have higher specificity than native ones. Modeling of mutant proteins showed that enhanced specificity is caused by increased complementarity between binding loop and the active site..")
35
Native state can be stabilized by reducing the difference in entropy between folded and unfolded conformations U F G Reaction coordinate ΔGΔG

36
Proteins can be made more stable by protein engineering. Three approaches to increase stability: Reduce the difference in entropy between folded and unfolded conformations Stabilize secondary structures Increase the number of hydrophobic interactions in the interior core

37
Model system: lysozyme from bacteriophage T4. Lysozyme has the ability to lyse certain bacteria by hydrolyzing the b-linkage between N-acetylmuramic acid (NAM) and N-acetylglucosamine (NAG) of the peptidoglycan layer in the bacterial cell wall. Conformational transition in lysozyme involves the relative movement of its two lobes to each other in a cooperative manner
 and N-acetylglucosamine (NAG) of the peptidoglycan layer in the bacterial cell wall. Conformational transition in lysozyme involves the relative movement of its two lobes to each other in a cooperative manner.")
38
Disulfide bridges increase protein stability. Increasing stability by reducing the number of unfolded conformations (since enthalpic contribution will be the same for folded and unfolded states). Task: to find positions on backbone where Cysteines can be introduced for disulfide bonds formation.
. Task: to find positions on backbone where Cysteines can be introduced for disulfide bonds formation..")
39
Strategy of introducing a new disulfide bond. B. Mathews, 1989: Analysis of disulfide bonds geometries in existing structures. Analysis of all pairs of amino acids which are close in space. Energy optimization of candidate disulfide bonds. Analysis of destabilizing effect of exchanging native amino acids into Cys. As a result: three disulfide bonds were introduced through mutagenesis experiments in lysozyme

40
Stability of mutants compared to wild- type protein. Measure of stability – melting temperature at which 50% of enzyme is inactivated during reversible heat denaturation. For wild-type Tm = 42 C. all mutants were more stable than wild-type. the longer the loop between Cys, the larger the effect (the more restricted is unfolded state). the more disulfide bonds were introduced, the more stable was the mutant. From B. Mathews et al
. the more disulfide bonds were introduced, the more stable was the mutant. From B. Mathews et al.")
41
Attempts to fill cavities to stabilize lysozyme failed… Introduction of cavities of size –CH 3 group destabilizes protein by ~ 1kcal/mol. T4 lysozyme has two cavities; mutations Leu Phe and Ala Val destabilize the protein by ~ 0.5-1.0 kcal/mol. New side-chains (Val and Phe) adopt unfavorable conformations in cavities.
 adopt unfavorable conformations in cavities..")
42
Classwork IV: analyzing the lysozyme’s mutants. Retrieve structure neighbors (1PQM and 1KNI) of 2LZM. Which mutant might have an increased stability and why?
 of 2LZM. Which mutant might have an increased stability and why .")
43
Can structural scaffolds be reduced in size with maintaining function? A.Braisted & J.A. Wells used Z-domain (58 residues) of bacterial protein A: removed third helix (truncated protein - 38 residues); mutated residues in the first and second helices; used phage display to select active forms; restored the binding of truncated protein.
 of bacterial protein A: removed third helix (truncated protein - 38 residues); mutated residues in the first and second helices; used phage display to select active forms; restored the binding of truncated protein..")
44
Designing an amino acid sequence that will fold into a given structure. Inverse protein folding problem: designing a sequence which will fold into a given structure – much easier than folding problem! B. Dahiyat & S. Mayo: designed a sequence of zinc finger domain that does not require stabilization by Zn. Wild type protein domain is stabilized by Zn (bound to two Cys and two His); mutant is stabilized by hydrophobic interactions.
; mutant is stabilized by hydrophobic interactions..")
45
Paracelsus challenge: convert one fold into another by changing 50% of residues. Challenge because all proteins with > 30% identity seem to have the same fold. L.Regan et al: Protein G (mainly beta-sheet) was converted to Rop protein (alpha-helical) by changing only 50% residues
 was converted to Rop protein (alpha-helical) by changing only 50% residues.")
Similar presentations
 and connectivity.>")

 evolution and other techniques.>")


![Structure Prediction. Tertiary protein structure: protein folding Three main approaches: [1] experimental determination (X-ray crystallography, NMR) [2]](/15/4859888/big_thumb.jpg "Structure Prediction. Tertiary protein structure: protein folding Three main approaches: [1] experimental determination (X-ray crystallography, NMR) [2]>")


![Structure Prediction. Tertiary protein structure: protein folding Three main approaches: [1] experimental determination (X-ray crystallography, NMR) [2]](/16/4993760/big_thumb.jpg "Structure Prediction. Tertiary protein structure: protein folding Three main approaches: [1] experimental determination (X-ray crystallography, NMR) [2]>")


![. Protein Structure Prediction [Based on Structural Bioinformatics, section VII]](/16/5116302/big_thumb.jpg ". Protein Structure Prediction [Based on Structural Bioinformatics, section VII]>")

 Today: Why bother about proteins/prediction Concepts of molecular.>")


 Why bother about proteins/prediction What is bioinformatics Protein databases Making use of database information –Predictions.>")


