Download presentation
Presentation is loading. Please wait.

1
Structure Prediction

2
Tertiary protein structure: protein folding Three main approaches: [1] experimental determination (X-ray crystallography, NMR) [2] Comparative modeling (based on homology) [3] Ab initio (de novo) prediction (Dr. Ingo Ruczinski at JHSPH)
![Tertiary protein structure: protein folding Three main approaches: [1] experimental determination (X-ray crystallography, NMR) [2] Comparative modeling (based on homology) [3] Ab initio (de novo) prediction (Dr.](http://images.slideplayer.com/16/4993760/slides/slide_2.jpg "Ingo Ruczinski at JHSPH).")
3
Experimental approaches to protein structure [1] X-ray crystallography -- Used to determine 80% of structures -- Requires high protein concentration -- Requires crystals -- Able to trace amino acid side chains -- Earliest structure solved was myoglobin [2] NMR -- Magnetic field applied to proteins in solution -- Largest structures: 350 amino acids (40 kD) -- Does not require crystallization
![Experimental approaches to protein structure [1] X-ray crystallography -- Used to determine 80% of structures -- Requires high protein concentration -- Requires crystals -- Able to trace amino acid side chains -- Earliest structure solved was myoglobin [2] NMR -- Magnetic field applied to proteins in solution -- Largest structures: 350 amino acids (40 kD) -- Does not require crystallization](http://images.slideplayer.com/16/4993760/slides/slide_3.jpg "Experimental approaches to protein structure [1] X-ray crystallography -- Used to determine 80% of structures -- Requires high protein concentration -- Requires crystals -- Able to trace amino acid side chains -- Earliest structure solved was myoglobin [2] NMR -- Magnetic field applied to proteins in solution -- Largest structures: 350 amino acids (40 kD) -- Does not require crystallization")
4
Steps in obtaining a protein structure Target selection Obtain, characterize protein Determine, refine, model the structure Deposit in database

5
X-ray crystallography http://en.wikipedia.org/wiki/X-ray_diffraction Sperm Whale Myoglobin

8
PDB April 08, 2008 – 50,000 proteins, 25 new experimentally determined structures each day New folds Old folds New PDB structures

9
Example 1wey

10
Ab initio protein prediction Starts with an attempt to derive secondary structure from the amino acid sequence – Predicting the likelihood that a subsequence will fold into an alpha- helix, beta-sheet, or coil, using physicochemical parameters or HMMs and ANNs – Able to accurately predict 3/4 of all local structures

11
Structure Characteristics

12
Beta Sheets

13
Ab Inito Prediction

14
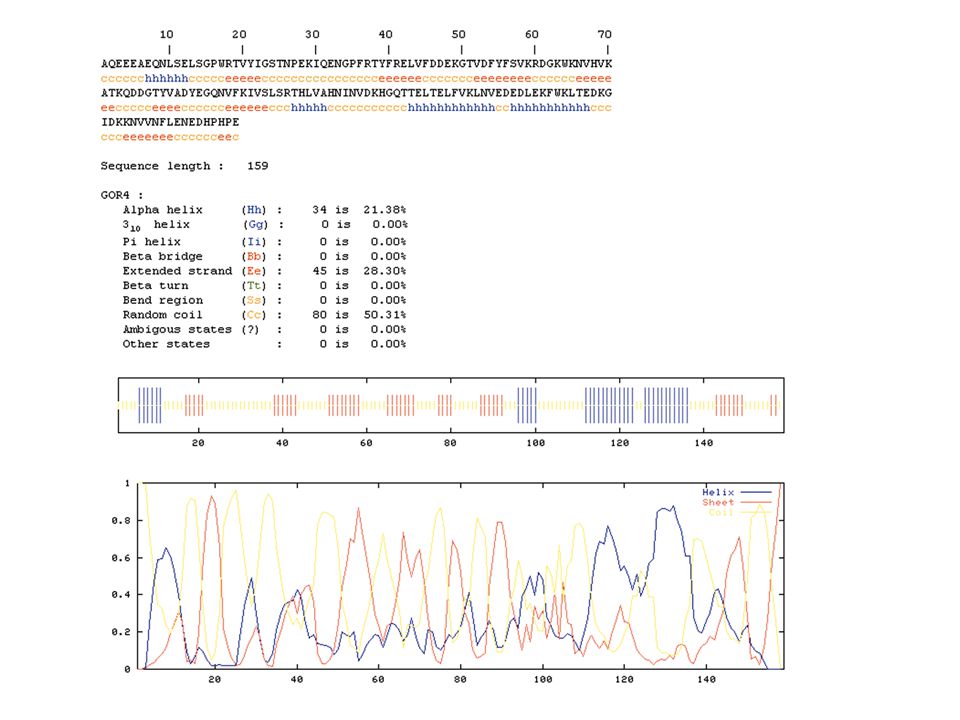
Secondary structure prediction Chou and Fasman (1974) developed an algorithm based on the frequencies of amino acids found in helices, -sheets, and turns. Proline: occurs at turns, but not in helices. GOR (Garnier, Osguthorpe, Robson): related algorithm Modern algorithms: use multiple sequence alignments and achieve higher success rate (about 70-75%) Page 279-280
: related algorithm Modern algorithms: use multiple sequence alignments and achieve higher success rate (about 70-75%) Page")
15
Table

19
Frequency Domain

20
Neural Networks

21
Training the Network Use PDB entries with validated secondary structures Measures of accuracy – Q 3 Score percentage of protein correctly predicted (trains to predicting the most abundant structure) – You get 50% if you just predict everything to be a coil – Most methods get around 60% with this metric
 – You get 50% if you just predict everything to be a coil – Most methods get around 60% with this metric")
22
Correlation Coeficient How correlated are the predictions for coils, helix and Beta-sheets to the real structures This ignores what we really want to get to – If the real structure has 3 coils, do we predict 3 coils? Segment overlap score (Sov) gives credit to how protein like the structure is, but it is correlated with Q 3
 gives credit to how protein like the structure is, but it is correlated with Q 3.")
23
Artificial Neural Network Predicts Structure at this point

24
Danger You may train the network on your training set, but it may not generalize to other data Perhaps we should train several ANNs and then let them vote on the structure

25
Profile network from HeiDelberg family (alignment is used as input) instead of just the new sequence On the first level, a window of length 13 around the residue is used The window slides down the sequence, making a prediction for each residue The input includes the frequency of amino acids occurring in each position in the multiple alignment (In the example, there are 5 sequences in the multiple alignment) The second level takes these predictions from neural networks that are centered on neighboring proteins The third level does a jury selection
 instead of just the new sequence On the first level, a window of length 13 around the residue is used The window slides down the sequence, making a prediction for each residue The input includes the frequency of amino acids occurring in each position in the multiple alignment (In the example, there are 5 sequences in the multiple alignment) The second level takes these predictions from neural networks that are centered on neighboring proteins The third level does a jury selection")
26
PHD Predicts 4 Predicts 6 Predicts 5

27
Fold recognition (structural profiles) Attempts to find the best fit of a raw polypeptide sequence onto a library of known protein folds A prediction of the secondary structure of the unknown is made and compared with the secondary structure of each member of the library of folds
 Attempts to find the best fit of a raw polypeptide sequence onto a library of known protein folds A prediction of the secondary structure of the unknown is made and compared with the secondary structure of each member of the library of folds")
28
Threading Takes the fold recognition process a step further: – Empirical-energy functions for residue pair interactions are used to mount the unknown onto the putative backbone in the best possible manner

29
Fold recognition by threading Query sequence Compatibility scores Fold 1 Fold 2 Fold 3 Fold N

30
CASP http://www.predictioncenter.org/casp8/index. cgi

31
SCOP SCOP: Structural Classification of Proteins. http://scop.mrc-lmb.cam.ac.uk/scop/

32
CATH CATH: Protein Structure Classification Class (C), Architecture (A), Topology (T) and Homologous superfamily (H) Class (C), Architecture (A), Topology (T) and Homologous superfamily (H)
, Architecture (A), Topology (T) and Homologous superfamily (H) Class (C), Architecture (A), Topology (T) and Homologous superfamily (H)")
Similar presentations










![Structure Prediction. Tertiary protein structure: protein folding Three main approaches: [1] experimental determination (X-ray crystallography, NMR) [2]](/15/4859888/big_thumb.jpg "Structure Prediction. Tertiary protein structure: protein folding Three main approaches: [1] experimental determination (X-ray crystallography, NMR) [2]>")

![Protein structure determination. Tertiary protein structure: protein folding Three main approaches: [1] experimental determination (X-ray crystallography,](/16/4956805/big_thumb.jpg "Protein structure determination. Tertiary protein structure: protein folding Three main approaches: [1] experimental determination (X-ray crystallography,>")


.>")

. Protein Folding Problem A protein folds into a unique 3D structure under physiological conditions Lysozyme sequence: KVFGRCELAA.>")

>")
 Protein Structure Prediction Protein Secondary Structure.>")


