Download presentation
Presentation is loading. Please wait.

1
Genboree Microbiome Workbench 16S Workshop Part I March 11 th, 2014 Julia Cope Emily Hollister Kevin Riehle

2
Genboree 16S Workshop Learning Objectives – Students should be able to take.sff files and user supplied information and produce: Metadata File PCoA Classification Distribution Expectations – Apply topics learned today before next meeting – Be able to discuss where issues arise – Be able to move knowledgeably through the whole Genboree Workflow

3
Genboree 16S Workshop Part II Learning Outcomes – Newer database version of RDP – How to take advantage? – Students should take user.sff files and user created metadata file and produce: (I can provide files if needed.) PCoA (QIIME) Classification Distribution (RDP) Expectations – Apply topics learned in tutorial – Be able to discuss where in the process issues arose – Have a hypothesis about your data issues if they happen
 PCoA (QIIME) Classification Distribution (RDP) Expectations – Apply topics learned in tutorial – Be able to discuss where in the process issues arose – Have a hypothesis about your data issues if they happen.")
4
Workshop Outline 16S Metadata File Genboree Workbench Workflow – Account – Group – Database – Project – Loading your files/samples/sequences (and linking) – QIIME – RDP – How to get help Wrap Up and Preparation for 2 nd Installment
 – QIIME – RDP – How to get help Wrap Up and Preparation for 2 nd Installment")
5
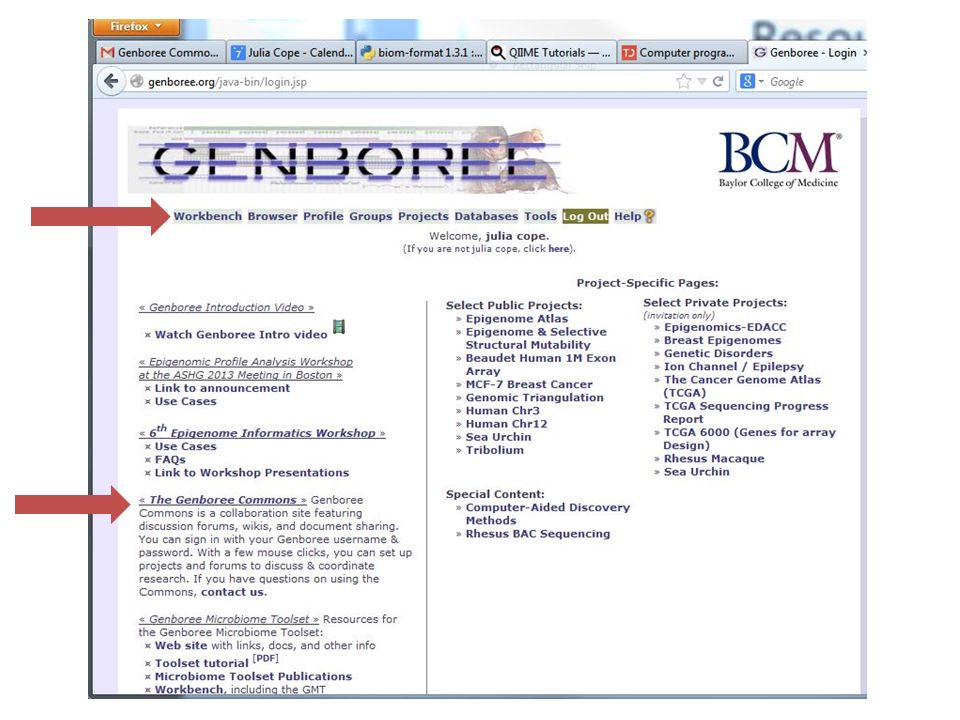
Resources Genboree Home Screen – http://genboree.org http://genboree.org Tutorials are located in the Genboree Commons – You must be signed in to open the following link – http://genboree.org/theCommons/projects/mw-march-2014 http://genboree.org/theCommons/projects/mw-march-2014 – Tutorial 1 Data Set: http://www.genboree.org/microbiome/include/data/tutorial_sequen ce_file.sff.gz http://www.genboree.org/microbiome/include/data/tutorial_sequen ce_file.sff.gz – Tutorial 2 Data Set: http://genboree.org/theCommons/attachments/3545/Tutorial_2.zip Projects are accessed through the Genboree Workbench

7
16S What is it? What part is being sequenced? – Here? – Elsewhere? How is this accomplished? – DNA to bead to light – Intro. to flow data and.sff file content – OUTPUT is an.sff file – Aside on zipping methods and large file transfers

8
Allmetrics.net Sales Material Tortoli E Clin. Microbiol. Rev. 2003;16:319-354 What is it? 16Svedberg (small sub-unit of the ribosome) What part is being sequenced? Here? - TCMC sequences the V5-V3 by 454 Elsewhere? - V3-V5, V1-V3, V9, V7-V9…many more. Know your variable regions 16S
 What part is being sequenced. Here. - TCMC sequences the V5-V3 by 454 Elsewhere. - V3-V5, V1-V3, V9, V7-V9…many more. Know your variable regions 16S.")
9
How is this accomplished? – DNA to bead to light http://cage.unl.edu/equipmentsoftware.shtml 454 Life Sciences Sales Materials

10
16S How is this accomplished? – DNA to bead to light http://cage.unl.edu/equipmentsoftware.shtml 454 Life Sciences Sales Materials

11
16S How is this accomplished? – DNA to bead to light – Intro to flow data and sff file content – OUTPUT is an.sff file – Standard Flowgram Format All reads are structured as linker-tag-primer Provides both identity and quality information http://cage.unl.edu/equipmentsoftware.shtml Allmetrics.net Sales Material

12
Genboree Workflow Take one step back from the Genboree Workflow and talk about input files. What do you do with your files? From: Genboree.org help files Meta- data.sff

13
Genboree Workflow What do you do with many files? Genboree takes.zip,.gzip,.txt, and.sff files – Compressed files are easier and faster to move – Multiple files are easier to move when compressed together in an archive Meta- data.sff.sff(s) should be archived and compressed. Meta data files are very small and do not need compression. Meta- data
 should be archived and compressed. Meta data files are very small and do not need compression. Meta- data.")
14
Metadata Files What data must you have? How should it be formatted for Genboree? What can you include? How to make it tab-delimited Include variable region or primer? Directional awareness on primers

15
Metadata Files What data must you have? – name – barcode – region or proximal & distal – First column must begin with # – #No_spaces_are_allowed_in_column_names_0123456789 How should it be formatted for Genboree? – Tab delimited What can you include? How to make it tab-delimited? Include variable region or primer? Directional awareness on primers

16
Metadata Files How to determine which to include - variable region or primers Directional awareness on primers Demo of making and saving as tab delimited #namebarcodeproximaldistalregionbody_site S_700033665CCGTTCCTCCCGTCAATTCMTTTRAGTCTGCTGCCTCCCGTAGGV3V5Stool S_700035861ACCGGCGTTCCCGTCAATTCMTTTRAGTCTGCTGCCTCCCGTAGGV3V5Stool S_700095543ACGAATTAACCCGTCAATTCMTTTRAGTCTGCTGCCTCCCGTAGGV3V5Stool S_700095850AACCGGATACCCGTCAATTCMTTTRAGTCTGCTGCCTCCCGTAGGV3V5Stool S_700101600AACGGAACGCCCGTCAATTCMTTTRAGTCTGCTGCCTCCCGTAGGV3V5Stool T_700016994AATAACCGTCCCGTCAATTCMTTTRAGTCTGCTGCCTCCCGTAGGV3V5Throat T_700095565TTAATGGAACCCGTCAATTCMTTTRAGTCTGCTGCCTCCCGTAGGV3V5Throat T_700095872CGGACCGGAACCCGTCAATTCMTTTRAGTCTGCTGCCTCCCGTAGGV3V5Throat T_700101388CCGAACGACCCGTCAATTCMTTTRAGTCTGCTGCCTCCCGTAGGV3V5Throat T_700101622TTCGTTCTTCCCGTCAATTCMTTTRAGTCTGCTGCCTCCCGTAGGV3V5Throat or

17
#namebarcodeproximaldistalregionbody_site S_700033665CCGTTCCTCCCGTCAATTCMTTTRAGTCTGCTGCCTCCCGTAGGV3V5Stool S_700035861ACCGGCGTTCCCGTCAATTCMTTTRAGTCTGCTGCCTCCCGTAGGV3V5Stool S_700095543ACGAATTAACCCGTCAATTCMTTTRAGTCTGCTGCCTCCCGTAGGV3V5Stool S_700095850AACCGGATACCCGTCAATTCMTTTRAGTCTGCTGCCTCCCGTAGGV3V5Stool S_700101600AACGGAACGCCCGTCAATTCMTTTRAGTCTGCTGCCTCCCGTAGGV3V5Stool T_700016994AATAACCGTCCCGTCAATTCMTTTRAGTCTGCTGCCTCCCGTAGGV3V5Throat T_700095565TTAATGGAACCCGTCAATTCMTTTRAGTCTGCTGCCTCCCGTAGGV3V5Throat T_700095872CGGACCGGAACCCGTCAATTCMTTTRAGTCTGCTGCCTCCCGTAGGV3V5Throat T_700101388CCGAACGACCCGTCAATTCMTTTRAGTCTGCTGCCTCCCGTAGGV3V5Throat T_700101622TTCGTTCTTCCCGTCAATTCMTTTRAGTCTGCTGCCTCCCGTAGGV3V5Throat Metadata Files - Demo Select the data above and Copy. Paste into Excel or an open source spreadsheet program. Be sure all entries are free of spaces and special characters and that all samples have the same number of columns. Avoid the column titles "state" and "type". Save As and select tab-delimited. Name your file in a clear and consistent manner. or

18
Metadata Files How to determine variable region vs. primer inclusion Directional awareness of primers If you aren’t sure, ask! What are these files often called: mapping, metadata, oligos, or linker-primer file. (Many others possible.) #namebarcodeproximaldistalregionbody_site S_700033665CCGTTCCTCCCGTCAATTCMTTTRAGTCTGCTGCCTCCCGTAGGV3V5Stool S_700035861ACCGGCGTTCCCGTCAATTCMTTTRAGTCTGCTGCCTCCCGTAGGV3V5Stool Allmetrics.net Sales Material
 #namebarcodeproximaldistalregionbody_site S_ CCGTTCCTCCCGTCAATTCMTTTRAGTCTGCTGCCTCCCGTAGGV3V5Stool S_ ACCGGCGTTCCCGTCAATTCMTTTRAGTCTGCTGCCTCCCGTAGGV3V5Stool Allmetrics.net Sales Material.")
19
Metadata Files Another example: Tutorial Set 2 Metadata What possible issues may arise with this metadata file? sampleNametagproximaldistalregionsample_periodtype Ferm_5AGCTTCGAGAGTTTGATCNTGGCTCAGCAGCMGCCGCNGTAANACV1V35Fermentation Ferm_2GCCATACATTGAGTTTGATCNTGGCTCAGCAGCMGCCGCNGTAANACV1V32Fermentation Ferm_3GCCAGCAAGTGAGTTTGATCNTGGCTCAGCAGCMGCCGCNGTAANACV1V33Fermentation Ferm_4CGTTAAGAGAGTTTGATCNTGGCTCAGCAGCMGCCGCNGTAANACV1V34Fermentation Ferm_1CTAACAGAGAGTTTGATCNTGGCTCAGCAGCMGCCGCNGTAANACV1V31Fermentation Soil_1ACGCAAAAGAGTTTGATCNTGGCTCAGCAGCMGCCGCNGTAANACV1V31Soil Soil_2CTAACTAAGAGTTTGATCNTGGCTCAGCAGCMGCCGCNGTAANACV1V32Soil Soil_3GCGACCTAGTGAGTTTGATCNTGGCTCAGCAGCMGCCGCNGTAANACV1V33Soil Soil_4AAGAATCAGAGTTTGATCNTGGCTCAGCAGCMGCCGCNGTAANACV1V34Soil Soil_5AGCGCAGAGAGTTTGATCNTGGCTCAGCAGCMGCCGCNGTAANACV1V35Soil

20
Metadata Files Another example What possible issues may arise with this metadata file? Change name => #name (or any #1 st entry) Change tag => barcode Change type => sample_type (do not name columns ‘type’ or ‘state’) Demo. making and saving as tab-delimited #namebarcode proximaldistalregionsample_period sample_type Ferm_5AGCTTCGAGAGTTTGATCNTGGCTCAGCAGCMGCCGCNGTAANACV1V35Fermentation Ferm_2GCCATACATTGAGTTTGATCNTGGCTCAGCAGCMGCCGCNGTAANACV1V32Fermentation Ferm_3GCCAGCAAGTGAGTTTGATCNTGGCTCAGCAGCMGCCGCNGTAANACV1V33Fermentation Ferm_4CGTTAAGAGAGTTTGATCNTGGCTCAGCAGCMGCCGCNGTAANACV1V34Fermentation Ferm_1CTAACAGAGAGTTTGATCNTGGCTCAGCAGCMGCCGCNGTAANACV1V31Fermentation Soil_1ACGCAAAAGAGTTTGATCNTGGCTCAGCAGCMGCCGCNGTAANACV1V31Soil Soil_2CTAACTAAGAGTTTGATCNTGGCTCAGCAGCMGCCGCNGTAANACV1V32Soil Soil_3GCGACCTAGTGAGTTTGATCNTGGCTCAGCAGCMGCCGCNGTAANACV1V33Soil Soil_4AAGAATCAGAGTTTGATCNTGGCTCAGCAGCMGCCGCNGTAANACV1V34Soil Soil_5AGCGCAGAGAGTTTGATCNTGGCTCAGCAGCMGCCGCNGTAANACV1V35Soil
 Change tag => barcode Change type => sample_type (do not name columns ‘type’ or ‘state’) Demo. making and saving as tab-delimited #namebarcode proximaldistalregionsample_period sample_type Ferm_5AGCTTCGAGAGTTTGATCNTGGCTCAGCAGCMGCCGCNGTAANACV1V35Fermentation Ferm_2GCCATACATTGAGTTTGATCNTGGCTCAGCAGCMGCCGCNGTAANACV1V32Fermentation Ferm_3GCCAGCAAGTGAGTTTGATCNTGGCTCAGCAGCMGCCGCNGTAANACV1V33Fermentation Ferm_4CGTTAAGAGAGTTTGATCNTGGCTCAGCAGCMGCCGCNGTAANACV1V34Fermentation Ferm_1CTAACAGAGAGTTTGATCNTGGCTCAGCAGCMGCCGCNGTAANACV1V31Fermentation Soil_1ACGCAAAAGAGTTTGATCNTGGCTCAGCAGCMGCCGCNGTAANACV1V31Soil Soil_2CTAACTAAGAGTTTGATCNTGGCTCAGCAGCMGCCGCNGTAANACV1V32Soil Soil_3GCGACCTAGTGAGTTTGATCNTGGCTCAGCAGCMGCCGCNGTAANACV1V33Soil Soil_4AAGAATCAGAGTTTGATCNTGGCTCAGCAGCMGCCGCNGTAANACV1V34Soil Soil_5AGCGCAGAGAGTTTGATCNTGGCTCAGCAGCMGCCGCNGTAANACV1V35Soil.")
21
7zip Zipping methods and large file transfers Compression and archiving of files Uncompressing in an easy to use format for PCs Demo compressing –.sff (s) – http://www.7-zip.org/ http://www.7-zip.org/ From: 7-zip.org
 – From: 7-zip.org")
22
Genboree Workflow Create Group Create Database Create Project Upload Files Create Samples (Sample Import using metadata file) Link Samples to Sequence Files (Sample File Linker) QC and Attach Sequences (Sequence Import) QIIME RDP
 Link Samples to Sequence Files (Sample File Linker) QC and Attach Sequences (Sequence Import) QIIME RDP ")
23
Genboree URL: http://www.genboree.orghttp://www.genboree.org Workbench and Commons Differences Account – How to create your account? – http://genboree.org/theCommons/ezfaq/show/p ublic-commons?faq_id=493 http://genboree.org/theCommons/ezfaq/show/p ublic-commons?faq_id=493 Workshop Home – http://genboree.org/theCommons/projects/mw- march-2014 http://genboree.org/theCommons/projects/mw- march-2014

25
Workbench Where is it? http://genboree.org/java-bin/workbench.jsphttp://genboree.org/java-bin/workbench.jsp Create a Group - Demo – Why? To serve as a project base – How to share it with others? – http://genboree.org/theCommons/ezfaq/show/public- commons?faq_id=494 http://genboree.org/theCommons/ezfaq/show/public- commons?faq_id=494 Create a Database - Demo – Why? To hold processed and pre-processed files – Using folders to organize the space – http://genboree.org/theCommons/ezfaq/show/public- commons?faq_id=491 http://genboree.org/theCommons/ezfaq/show/public- commons?faq_id=491 Create a Project - Demo – Why? To have a record of the major level processes that you have used on your data – Importance of tracking information for multiple users in a group – http://genboree.org/theCommons/ezfaq/show/public- commons?faq_id=492 http://genboree.org/theCommons/ezfaq/show/public- commons?faq_id=492

26
Genboree Workflow Create Group Create Database Create Project Upload Files Create Samples (Sample Import using metadata file) Link Samples to Sequence Files (Sample File Linker) QC and Attach Sequences (Sequence Import) QIIME RDP
 Link Samples to Sequence Files (Sample File Linker) QC and Attach Sequences (Sequence Import) QIIME RDP ")
27
Upload Files What to import (upload) – Meta data –.sff (s) – Can both meta data and sffs be in one file? No - upload them separately..sffs will need unpacking while meta data files will need converting. Shortcutting this step can cause odd problems down the line. Importing files and choosing to extract will cause the system to queue the process. The process may take a few moments. Now that I have it uploaded…How to edit and remove files? - Demo

28
Genboree Workflow Create Group Create Database Create Project Upload Files Create Samples (Sample Import using metadata file) Link Samples to Sequence Files (Sample File Linker) QC and Attach Sequences (Sequence Import) QIIME RDP
 Link Samples to Sequence Files (Sample File Linker) QC and Attach Sequences (Sequence Import) QIIME RDP ")
29
Create Samples (Import) Import samples singly or in multiples – Creating and adding samples to a set – Import Behavior – Assign samples to a set What is a sample set? – Why use them? Grouping for downstream analysis Makes Genboree use faster on user (don’t have to move each file around) Editing sample information
 Editing sample information.")
30
Create Samples (Import) Import samples singly or in multiples: Demo – Creating and adding samples to a set Input Window: Metadata file Output Window: Target Database Data> Samples & Sample Sets> Samples> Import Samples Double check your Input, Target, and Settings – Import Behavior – Create New Record – Keep Existing – Merge and Update Use this one by default – Replace Existing – Assign Samples to new Sample Set Name the folder or leave blank to not create a set Can be added to a set later
 Import samples singly or in multiples: Demo – Creating and adding samples to a set Input Window: Metadata file Output Window: Target Database Data> Samples & Sample Sets> Samples> Import Samples Double check your Input, Target, and Settings – Import Behavior – Create New Record – Keep Existing – Merge and Update Use this one by default – Replace Existing – Assign Samples to new Sample Set Name the folder or leave blank to not create a set Can be added to a set later")
31
Create Samples (Import) What is a sample set? – Why use them? Grouping for downstream analysis Makes Genboree use faster on user (don’t have to move each file around) Editing sample information – What isn’t possible (right now)? Editing column titles Adding single samples de novo
 Editing sample information – What isn’t possible (right now). Editing column titles Adding single samples de novo.")
32
Sample Set Management Demo. adding samples to a sample set – Input Window: Sample to be added – Output Window: Target Sample Set – Data> Samples & Sample Sets> Sample Sets> Add Sample to Sample Set Demo. editing Sample (or Sample Set) data – Input Window: Sample to be edited – Output Window: Blank – Data> Samples & Sample Sets> Samples> Edit Samples This is important for later stages – Makes Sequence Import easier and cleaner
 data – Input Window: Sample to be edited – Output Window: Blank – Data> Samples & Sample Sets> Samples> Edit Samples This is important for later stages – Makes Sequence Import easier and cleaner.")
33
Sample Set Management Editing Sample (or Sample Set) data – Move boxes before saving or you will lose your edit.
 data – Move boxes before saving or you will lose your edit.")
34
Genboree Workflow Create Group Create Database Create Project Upload Files Create Samples (Sample Import using metadata file) Link Samples to Sequence Files (Sample File Linker) QC and Attach Sequences (Sequence Import) QIIME RDP
 Link Samples to Sequence Files (Sample File Linker) QC and Attach Sequences (Sequence Import) QIIME RDP ")
35
Link Samples to Sequence Files Sample file linker tool – The name is opposite the file positions required. Arrangement in the Input Window: –.sff Sample Set or –.sff Sample –.sff Sample –.sff Sample Output Window: Empty Demo. how to do it and how to check it has been done.

36
Link Samples to Sequence Files How to check your linked files? – The prompt screen on linking – The e-mail when complete – The Sample Edit tool – look for fileLocation column. – Demo. looking at linked fileLocation Input Window: Sample to be edited Output Window: Blank Data> Samples & Sample Sets> Samples> Edit Samples

37
Genboree Workflow Create Group Create Database Create Project Upload Files Create Samples (Sample Import using metadata file) Link Samples to Sequence Files (Sample File Linker) QC and Attach Sequences (Sequence Import) QIIME RDP
 Link Samples to Sequence Files (Sample File Linker) QC and Attach Sequences (Sequence Import) QIIME RDP ")
38
Sequence Import Choose one or more samples to load sequences – Input Window: Sample(s) or Sample Set – Output Window: Target Database – Metagenome> Data Initialization> Import 16S rRNA Sequences Check quality of import Fixing the files when something has gone wrong – When it is possible? – When to start over? Download files from Genboree

39
Sequence Import Choose one or more samples to load sequences – Demo. – Input Window: Sample(s) or Sample Set – Output Window: Target Database – Metagenome> Data Initialization> Import 16S rRNA Sequences
 or Sample Set – Output Window: Target Database – Metagenome> Data Initialization> Import 16S rRNA Sequences.")
40
Sequence Import Check quality of import

41
Sequence Import Fixing the files when something has gone wrong

42
Sequence Import Fixing the files when something has gone wrong – When it is possible? Bad barcode? Sample info. wrong? – Primers – Region – Direction Bad file? – When to start over?

43
Sequence Import Download files from Genboree Click on file In Details Window, choose Download Start with – sequences_metrics_ summary.xls – Easy to open – No compression

44
Sequence Import When problems arise, check the: – sample.metadata – Does it match what you put in? – fasta.result.tar.gz – Look at the.fasta files See barcodes See primers Notepad for metadata Bioedit to open fasta – Use WINE on Mac

45
Genboree Workflow Create Group Create Database Create Project Upload Files Create Samples (Sample Import using metadata file) Link Samples to Sequence Files (Sample File Linker) QC and Attach Sequences (Sequence Import) QIIME RDP
 Link Samples to Sequence Files (Sample File Linker) QC and Attach Sequences (Sequence Import) QIIME RDP ")
46
Break

47
Genboree Workflow Create Group Create Database Create Project Upload Files Create Samples (Sample Import using metadata file) Link Samples to Sequence Files (Sample File Linker) QC and Attach Sequences (Sequence Import) QIIME RDP
 Link Samples to Sequence Files (Sample File Linker) QC and Attach Sequences (Sequence Import) QIIME RDP ")
48
Data Analysis - QIIME How to select samples for analysis Chimera removal and why you should be thinking about it Output – downloading and organization – making sense of the files

49
Data Analysis - QIIME How to select samples for analysis

50
Data Analysis - QIIME – Selecting samples for analysis INPUT = One or more Sequence Import folders – All should be of the same variable region; ideally produced with the same primer and sequencing direction OUTPUT Targets = Your database (required), your project (optional)
, your project (optional)")
51
Data Analysis - QIIME Caveats: All samples in your input folder will be analyzed – This includes no-template controls and positive controls – The % variation explained by you PCoA may be influenced by the inclusion of these samples QIIME on Genboree is not currently set up to allow users to subsample their data – This can be problematic if sequencing depth varies substantially across samples – It does however perform a “rounding up” normalization step

52
A bit about sequencing depth How deep should you go? There is no good answer Strong biological patterns can be detected with low sequencing depth – 10s to 100s of sequences can sometimes be enough – 1000s tend to be the norm Subtle biological patterns tend to require greater sequencing depth for detection Sequencing depth can be dictated by: – Sample quality – The number of samples placed on a run – Project budget Kuczynzski et al. 2010 Nature Methods 7: 813-819

53
Unequal sequencing depth What’s the problem? http://www.cs.unc.edu/~lguan/Research.files/backgroundSubtractionResult.JPG Being certain that you are seeing the full view (…or at least equivalent glimpses of the) of your communities
 of your communities.")
54
Unequal sequencing depth What’s the problem? Unequal depth Avg Red = 5995 seqs Avg Blue = 11672 seqs Same data set Sampled are colored by library size Red ~4000 Orange ~5000 Yellow ~6000 Green 8,000-10,000 Blues 11,000-17,000

55
Unequal sequencing depth What’s the problem? Unequal depth Avg Red = 5995 seqs Avg Blue = 11672 seqs Equal depth All libraries were sub-sampled to ~4000 reads.

56
Data Analysis - QIIME Chimera removal and why you should be thinking about it – What is a chimeric sequence? – How frequently do they occur? – An example from real data – Why should you think about chimeras? – How to screen for chimeras using Genboree

57
What is a Chimeric Sequence? – In Greek mythology: A creature that was an amalgam of multiple animals Body of a lion, head of a goat, tail resembling a snake – In your sequence data: The combination of multiple sequences during PCR to create a hybrid – In sequence databases: A not-so-small nightmare of junk data Mis-annotation Enhanced “discovery” of novel organisms Chimera generation figure from: Haas et al. 2011, Genome Research 21:494-504

58
How frequently do chimeras occur? – Schloss et al 2011: With mock communities of known composition: ~8% of raw sequences were chimeric Incidence increased with sequencing depth – Approaches for detection: Multiple algorithms available Genboree uses ChimeraSlayer – How it works: The ends of each read (~30% of total length) are compared to a chimera-free reference database Potential “parent” sequences are identified Identity of potential chimera to in silico chimera evaluated Schloss et al. 2011 PLoS ONE 6(12):e27310 AATCGCGACCTGTTTAACCGTAGGTC AAACGCTTACGGAGCTACACGAGTC Query Parent 1 Parent 2 AATCGCGACCTGTGCTACACGGGTA AATCGCGACCTGTTTAACCGTAGGTC AAACGCTTACGGAGCTACACGGGTA Query Parent 1 Parent 2 Likely Chimera Non-chimera
 are compared to a chimera-free reference database Potential parent sequences are identified Identity of potential chimera to in silico chimera evaluated Schloss et al PLoS ONE 6(12):e27310 AATCGCGACCTGTTTAACCGTAGGTC AAACGCTTACGGAGCTACACGAGTC Query Parent 1 Parent 2 AATCGCGACCTGTGCTACACGGGTA AATCGCGACCTGTTTAACCGTAGGTC AAACGCTTACGGAGCTACACGGGTA Query Parent 1 Parent 2 Likely Chimera Non-chimera.")
59
An example from real data Chimeric alignment from: Haas et al. 2011, Genome Research 21:494-504 Alignment of chimeric sequences derived from Streptococcus (top, red) and Staphylococcus (bottom, black) Sequences were generated from 4 replicate PCR reactions/454 runs of V3V5 sequence
 and Staphylococcus (bottom, black) Sequences were generated from 4 replicate PCR reactions/454 runs of V3V5 sequence.")
60
Why should you think about chimeras? – Spurious results Artificially increases estimates of richness and diversity You may discover a “new” (but fake) species – Should you trust all flagged chimeras? Most people do but….buyer beware False-positive rates are in the 1-4% range Some taxa are poorly represented in reference databases Prevotella and Acinetobacter are known to produce false-positive results in ChimeraSlayer – How to verify (digging in to your QIIME output) Obtain representative sequence(s) and verify their identity (e.g., BLAST vs. NCBI nt database, RDP SeqMatch) Sogin et al 2006 PNAS 103:12115-12120
 species – Should you trust all flagged chimeras. Most people do but….buyer beware False-positive rates are in the 1-4% range Some taxa are poorly represented in reference databases Prevotella and Acinetobacter are known to produce false-positive results in ChimeraSlayer – How to verify (digging in to your QIIME output) Obtain representative sequence(s) and verify their identity (e.g., BLAST vs. NCBI nt database, RDP SeqMatch) Sogin et al 2006 PNAS 103:")
61
How to screen chimeras in Genboree – Run a QIIME job INPUT = Sequence Import folder OUTPUT Targets = Your database (required), your project (optional)
, your project (optional)")
62
How to screen chimeras in Genboree – Select “Remove Chimeras” in the Tool Settings dialogue box Provide a study name Provide a job name (TIP: add chimeras_removed to you job name so that your output reflects that you selected this option) Click SUBMIT
 Click SUBMIT")
63
Data Analysis - QIIME Output – downloading and organization – making sense of the files

64
How do I get my files out? – Entire folders can be archived/downloaded INPUT = Folder to be archived OUTPUT = Database to house archive

65
How do I get my files out? – Entire folders can be archived/downloaded Provide and archive name Choose your compression type Decide if you want the directory structure to be preserved SUBMIT

66
How do I get my files out? – Single files, including archives, can be downloaded one by one Click on your file of interest in the DATA SELECTOR window Click on the “Click to Download File” link in the DETAILS window Save the file to your computer or storage drive Most file types will require decompression

67
QIIME – making sense of the files – fasta.result.tar.gz – jobFile.json – mapping.txt – otu.table – phylogenetic.result.tar.gz – plots.result.tar.gz – raw.results.tar.gz – repr_set.fasta.ignore – sample.metadata – settings.json – taxonomy.result.tar.gz

68
QIIME – making sense of the files – fasta.result.tar.gz: multiple sequence alignment of your representative sequences file. Rep seqs = representative sequence for each OTU. – jobFile.json: a log of the settings used by Genboree to run your analysis – mapping.txt: a QIIME-compatible metadata file, includes barcode information – otu.table: a spreadsheet of OTU by sample distributions – phylogenetic.result.tar.gz: a phylogenetic tree of your rep seqs, additional files required for iTOL – plots.result.tar.gz: figures, html files for all PCoA plots produced in your QIIME run – raw.results.tar.gz: mapping file, otu table, rep seqs file, distance matrices underlying all PCoA calculations – repr_set.fasta.ignore: RDP classification (with confidence scores) of each rep seq – sample.metadata: like the mapping.txt file, with additional file locations for Genboree – settings.json: similar to the jobFile.json file – taxonomy.result.tar.gz: taxonomic summaries (per sample, at the Kingdom, Phylum, Class, Order, Family, and Genus levels)
 of each rep seq – sample.metadata: like the mapping.txt file, with additional file locations for Genboree – settings.json: similar to the jobFile.json file – taxonomy.result.tar.gz: taxonomic summaries (per sample, at the Kingdom, Phylum, Class, Order, Family, and Genus levels).")
69
Genboree Workflow Create Group Create Database Create Project Upload Files Create Samples (Sample Import using metadata file) Link Samples to Sequence Files (Sample File Linker) QC and Attach Sequences (Sequence Import) QIIME RDP
 Link Samples to Sequence Files (Sample File Linker) QC and Attach Sequences (Sequence Import) QIIME RDP ")
70
Data Analysis - RDP How to select samples Output – Downloading and organization – making sense of the files

71
Data Analysis - RDP – Selecting samples for analysis INPUT = One or more Sequence Import folders – All should be of the same variable region; ideally produced with the same primer and sequencing direction OUTPUT Targets = Your database (required), your project (optional)
, your project (optional)")
72
Data Analysis - RDP Caveats: All samples in your input folder will be analyzed – This includes no-template controls and positive controls RDP on Genboree does not pre-filter for chimeric sequences RDP on Genboree is not currently set up to allow users to subsample their data – Depending on your application, this may be problematic if sequencing depth varies substantially across samples – It does however perform a “rounding up” normalization step and presents data on a relative abundance basis

73
How do I get my files out? – Entire folders can be archived/downloaded INPUT = Folder to be archived OUTPUT = Database to house archive

74
How do I get my files out? – Entire folders can be archived/downloaded Provide and archive name Choose your compression type Decide if you want the directory structure to be preserved SUBMIT

75
How do I get my files out? – Single files, including archives, can be downloaded one by one Click on your file of interest in the DATA SELECTOR window Click on the “Click to Download File” link in the DETAILS window Save the file to your computer or storage drive Most file types will require decompression

76
RDP – making sense of the files – domain.result.tar.gz – phylum.result.tar.gz – class.result.tar.gz – order.result.tar.gz – family.result.tar.gz – genus.result.tar.gz – sample.metadata – settings.json – count.result.tar.gz – count.xlsx – count_normalized.xlsx – weighted.xlsx – weighted_normalized.xlsx – png.result.tar.gz

77
RDP – making sense of the files – domain.result.tar.gz – phylum.result.tar.gz – class.result.tar.gz – order.result.tar.gz – family.result.tar.gz – genus.result.tar.gz – sample.metadata – settings.json – count.xlsx – count_normalized.xlsx – weighted.xlsx – weighted_normalized.xlsx – png.result.tar.gz Per sample summaries at various taxonomic levels, including raw counts and weighted values Per sample summaries at various taxonomic levels, raw counts or relative abundances (normalized) All of the plots produced during your run (e.g., heatmaps, stacked bar graphs) Per sample summaries at various taxonomic levels, weighted by confidence of ID assignments (raw counts or normalized)
 All of the plots produced during your run (e.g., heatmaps, stacked bar graphs) Per sample summaries at various taxonomic levels, weighted by confidence of ID assignments (raw counts or normalized)")
78
Individual Time Confirm user accounts are created. Confirm users know where mock data or their data set are.

Similar presentations







Version Control (SVN).>")












