Download presentation
Presentation is loading. Please wait.

1
ABNORMALITIES IN DERMAL CONNECTIVE TISSUE
Chapter 25

2
Elastosis perforans serpiginosa EPS
Skin colored keratotic papules, 2-5 mm, confluently grouped in a serpiginous or horseshoe-shaped arrangement Typically occur on the neck, may involve other sites including the face, arms, legs Disseminated lesions occur in Down syndrome MC in young adults, M:F 4:1

3
Elastosis perforans serpiginosa EPS
Runs a variable course with spontaneous resolution at 6 mo to 5 yrs Atrophic scar often remains Frequent assoc conditions include Down syndrome, Ehlers-Danlos syndrome, osteogenesis imperfecta, Marfan syndrome, Rothmund-Thomson syndrome, acrogeria, systemic sclerosis

4
Elastosis perforans serpiginosa EPS
Distinctive histopathologic changes Elongated tortuous channels in the epidermis into which elastic tissue perforates and is excruded Treatment is difficult LN2

5
Elastosis perforans serpiginosa EPS, penicillamine

6
Keratotic papules of EPS penicillamine TX

7
Annular plaques of EPS

8
EPS Hyperelastic epidermis clutches the increased dermal elastic fibers like a claw

9
EPS Transepidermal elimination of neutrophils and elastic fibers from the dermis through a channel in the epidermis

10
Reactive perforating collagenosis RPC
Rare, familial, nonpruritic skin disorder Papules on the extremities face or buttocks Lesions begin in 2nd decade Involution after 6-8 weeks, with new crops for years May be a reaction to connective tissue injury acquired form, may be assoc. with systemic disease, TX – underlying disease

11
Keratotic papule of RPC
Lesion followed minor trauma on the upper extremity

12
Pseudoxanthoma elasticum PXE
Inherited skin disorder involving the connective tissue of the skin, eye, and cardiovascular system Recessive and dominant inheritance Small, circumscribed, yellowish or cream-colored, creplike, lax, redundant folds, flecked with yellow papules, “plucked chicken skin” Characteristic exaggerated nasolabial folds

13
Pseudoxanthoma elasticum PXE
Characteristic retinal change is the angioid streak, 85% of PXE pts have retinal findings May be the only sign of disease for years Involvement of the cardiovascular system occurs with a propensity to hemorrhage The vascular events are caused by the degeneration of the elastic fibers in the vascular media

14
Clinical features of PXE
Yellowish papules, calcified plaques, sagging skin

15
Mucosal lesions Angioid streaks

16
Pseudoxanthoma elasticum PXE
Mitral valve prolapse, 71% of 14 pts Any patient with hypertension at a young age should be examined for the PXE Histologically, throughout the mid-dermis the elastic fibers are swollen and fragmented or granular “raveled wool” No distinctive therapy is available Progressive loss of vision, limit dietary calcium and phosphorus

17
Histopathology of PXE A. calcium deposits on elastic fibers in advanced PXE B. irregularly clumped elastic fibers, Verhoeff van Giesson

18
Perforating calcific elastosis
Periumbilical perforating PXE, and localized acquired cutaneous PXE Acquired, localized cutaneous disorder, most frequently found in the obese, multiparous, middle-aged women Yellowish, lax, well circumscribed, reticulated or cobblestones plaques occur in the periumbilical region with keratotic surface papules

19
Perforating calcific elastosis
A distinct disorder that shares some features with PXE, without systemic features It is suggested that repeated trauma of pregnancy, obesity or surgery promotes elastic fiber degeneration No effective therapy

20
Ehlers-Danlos syndromes
Cutis hyperelastica, India rubber skin, and elastic skin A group of genetically distinct connective tissue disorders characterized by excessive stretchability and fragility of the skin Tendency toward easy scar formation, calcification of the skin to produce, pseudotumors, and hyperextensibility of the joints

24
Two types of growths seen with EDS
Molluscum pseudotumor, a soft fleshy nodule seen in areas of trauma Spheroids, hard subcutaneous nodules that become calcified, ? Result of fat necrosis Special features associated with various subtypes

25
Types I, II, III and one subtype each of types of IV, VII and possibly VIII, AD
One subtype of IV, VI, VII, and X, AR Type V, X-linked inheritance Treatment is supportive Avoidance of trauma

26
Clinical features of Ehlers-Danlos syndrome

27
Marfan syndrome Disorder of connective tissue transmitted as an AD trait Skeletal, cardiovascular, and ocular involvement, it is one of the more common inherited diseases Important abnormalities include tallness, loose-jointedness, a dolichocephalic skull, high arched palate, arachnodactyly, pigeon breast, pes planus, poor muscular tone, large deformed ears

28
Ascending aortic aneurysm and mitral valve prolapse are commonly seen
Ectopic lentis and striae, EPS rarely Gene defect localized to chromosome 15 Abnormal elastic tissue in fibrillin 1 and fibrillin 2

29
Cutis laxa (generalized elastosis)
Dermatomegaly, dermatolysis, chalazoderma and pachydermatocele Characterized by loose, redundant skin, hanging in folds Drooping skin around the eyelids, cheeks and neck, bloodhound-like facies Usually entire integument is involved AD, primarily cutaneous, good prognosis AR, significant internal involvement, die young

30
Clinical features of cutis laxa

31
The X-linked recessive cutis laxa, occipital horn syndrome
Nonfamilial forms have been described Some cases have been associated with an underlying disease or a preceding inflammatory skin process mid-dermal elastosis is an acquired, nonfamilial condition affecting primarily young women, cause unknown

32
Treatment has been generally disappointing
Multiple surgical procedures have been largely unsuccessful

33
Cutis laxa (generalized elastosis)
Premature aging, severe pulmonary emphysema, and fragmentation of dermal elastic fibers

34
Blepharochalasis The eyelid skin becomes so lax that it falls in redundant folds over the lid margin Uncommon, occurs in young people at time of puberty Recurrent transitory swelling of the lids leads to stretching Usually bilateral, generally sporadic, AD Lack of elastic fibers, and abundant IgA deposits have been demonstrated

35
Blepharochalasis Ascher syndrome consist of progressive enlargement of the upper lip and blepharochalasis, treatment is surgical

36
Anetoderma (macular atrophy)
A group of disorders characterized by looseness of the skin, are due to loss of elastic tissue without apparent changes in the skin

37
Anetoderma (macular atrophy)
")
39
Clinical findings in both primary and secondary anetoderma are 5 – 10 mm atrophic plaques that are well defined Lesions protrude through the skin and on palpation have less resistance than surrounding skin, “button-hole” sign Trunk, shoulders, upper arms, and thighs Anetoderma of prematurity

40
Anetoderma (macular atrophy)
")
41
Anetoderma, decrease of the elastic fibers in the papillary and reticular dermis

42
Striae distensae Striae atrophicae
Depressed lines or bands of thin, reddened skin Become white, smooth, shiny and depressed Can occur during or after pregnancy, on the breasts, after sudden weight gain or muscle mass Cushing’s syndrome either endogenous or induced Prolonged application of topical steroids

43
Striae distensae Overtime striae become less noticeable
Topical tretinoin, and vascular lasers

44
Striae rubra, striae alba

45
Linear focal elastosis (elastotic striae)
Asymptomatic, palpable, striaelike yellow line of the middle and lower back Distinguished from striae in that the linear bands are not elevated, not depressed, and yellow, not pink or white

46
Acrodermatitis chronica atrophicans
Acquired diffuse thinning of the skin begins with an early reddish appearance on extensor surfaces Progresses to smooth , soft, atrophic skin Results from infection with Borrelia

47
Osteogenesis imperfecta
Lobstein’s syndrome Affects the bones, joints, eyes, ears, and skin types I-IV, I and IV AD, II and III AD/AR 50% are type I, type II is lethal within 1st week of life Brittle bones, fractures occur early in life, sometimes in utero Loose-jointedness and dislocations

48
Osteogenesis imperfecta
Blue sclera may be a valuable diagnostic clue, minimal functional importance Deafness develops in many by 2nd decade The skin is thin and rather translucent and healing wounds result in spreading atrophic scars EPS has been described

49
Osteogenesis imperfecta
The basic defect is abnormal collagen synthesis, resulting in type I collagen of abnormal structure The major causes of death are respiratory failure secondary to severs kyphoscoliosis and head trauma, mostly observed in type III Type I and IV have a normal life span TX – Pamidronate encouraging

50
homocystinuria An inborn error in the metabolism if methionine
Characterized by the presence of homocystine in the urine and systemic abnormalities of the connective tissue cystathionine synthetase enzyme deficient Genu valgum, kyphoscoliosis, pigeon breast, frequent fractures

51
homocystinuria Facial skin has a characteristic flush, malar
Other skin is blotchy red, suggestive of livedo reticularis Hair is fine, sparse and blonde Teeth are irregularly aligned Downward dislocations of lens TX – hydroxocobalamin and cyanocobalamin, variable results

52
ERRORS IN METABOLISM Chapter 26

53
SYSTEMIC AMYLOIDOSIS primary systemic amyloidosis
Involves mesenchymal tissue, the tongue, heart, gastrointestinal, and skin Cutaneous manifestations in 40% The amyloid fibril proteins are composed of protein AL Derived from immunoglobulin light chains 90% will have fragment in urine and serum

56
SYSTEMIC AMYLOIDOSIS primary systemic amyloidosis
The cutaneous eruption usually begins as shiny, smooth, firm, flat-topped, or spherical papules of waxy color, may appear translucent Lesions coalesce to form nodules and plaques Eyes, nose, mouth, and mucocutaneous junctions, areas commonly involved Purpuric lesions and ecchymosis occur 15% Most common cutaneous manifestation Results from amyloid infiltration of vessels

57
SYSTEMIC AMYLOIDOSIS primary systemic amyloidosis
Glossitis, with macroglossia, occurs in at least 20 % May be an early symptom, can cause dysphagia Tongue becomes enlarged with indentations from teeth Bullous disease is rare, heals with scarring Subepidermal, DDX PCT and EBA

58
SYSTEMIC AMYLOIDOSIS primary systemic amyloidosis
May present with many systemic findings, carpel tunnel syndrome, other peripheral neuropathies, arthropathy, GI bleeding, cardiac disease Prognosis is poor, median survival 13 mo, 5 in myeloma associated cases Treatment is difficult, melphalen, prednisone, hematopoietic stem cell transplantation

59
primary systemic amyloidosis
Macroglossia with dental impression of the tongue

60
primary systemic amyloidosis
Periorbital ecchymosis, “raccoon sign”

61
primary systemic amyloidosis
Numerous waxy and translucent papules

62
Secondary systemic amyloidosis
Amyloid involvement of the adrenals, liver spleen, and kidney as a result of some chronic disease, such as, tuberculosis, lepromatous leprosy and others Skin is not involved Amyloid fibrils are designated AA, protein component is unrelated to immunoglobulin Treat the underlying condition

63
CUTANEOUS AMYLOIDOSIS primary cutaneous amyloidosis
Divided into macular and lichen amyloid, most patients have only one form Asian , Hispanic, and Middle Eastern are predisposed The deposited amyloid material contains keratin as its protein Histologic picture is similar for both forms Differ only in size of amyloid deposits Absence of amyloid deposits around blood vessels excludes systemic involvement

64
Macular amyloidosis Typical cases exhibit moderately pruritic, brown, rippled macules Characteristically located on the interscapular region of the back Lack of uniformity gives “salt and pepper” appearance May involve other areas A chronic condition

65
Hyperpigmentation of macular amyloidosis with the characteristic rippled pattern

66
Lichen amyloidosis Characterized by the appearance of paroxysmally itchy lichenoid papules, typically appearing bilaterally on the shins Small, brown , discrete, slightly scaly papules Group to form large plaques Treatment is frequently unsatisfactory High potency corticosteroids, oral retinoids, cyclophosphamide, dermabrasion and occlusion

67
Lichen amyloidosis Keratotic, hyperigmented plaques on the legs

68
Nodular amyloidosis A rare form of primary localized cutaneous amyloidosis Single, or rarely, multiple nodules found Extremities, trunk, genitals and face Overlying epidermis may resemble large bullae Lesions contain numerous plasma cells, amyloid is immunoglobulin-derived AL TX – physical removal or destruction

70
Secondary cutaneous amyloidosis
Following PUVA therapy and in benign and malignant cutaneous neoplasms, deposits of amyloid may be found Most frequently associated neoplasms are NMSC and seborrheic keratosis In all cases, this is keratin-derived amyloid

71
Familial syndromes associated with amyloidosis (heredofamilial amyloidosis)
Familial syndromes have been reported that have either systemic or localized amyloidosis Muckle-Wells syndrome Multiple endocrine neoplasia IIA

72
Familial syndromes associated with amyloidosis (heredofamilial amyloidosis)
Most forms present with neurologic disease and are now designated familial amyloidotic polyneuropathy Four types identified FAP I through IV AD inherited

73
PORPHYRIAS Porphyrinogens are the building blocks of all the hemoproteins Produced primarily in the liver, bone marrow and erythrocytes Each form of porphyria is associated with a deficiency in the metabolic pathway of heme synthesis Photosensitivity may be seen in some types

77
Absorption of UV radiation in the Soret band (400-410 nm) by the increased porphyrins
Activated porphyrins are unstable and transfer energy as the return to their ground state. A reactive oxygen species is created causes tissue damage

78
Current grouping of the porphyrias is based on the primary site of increased porphyrin production
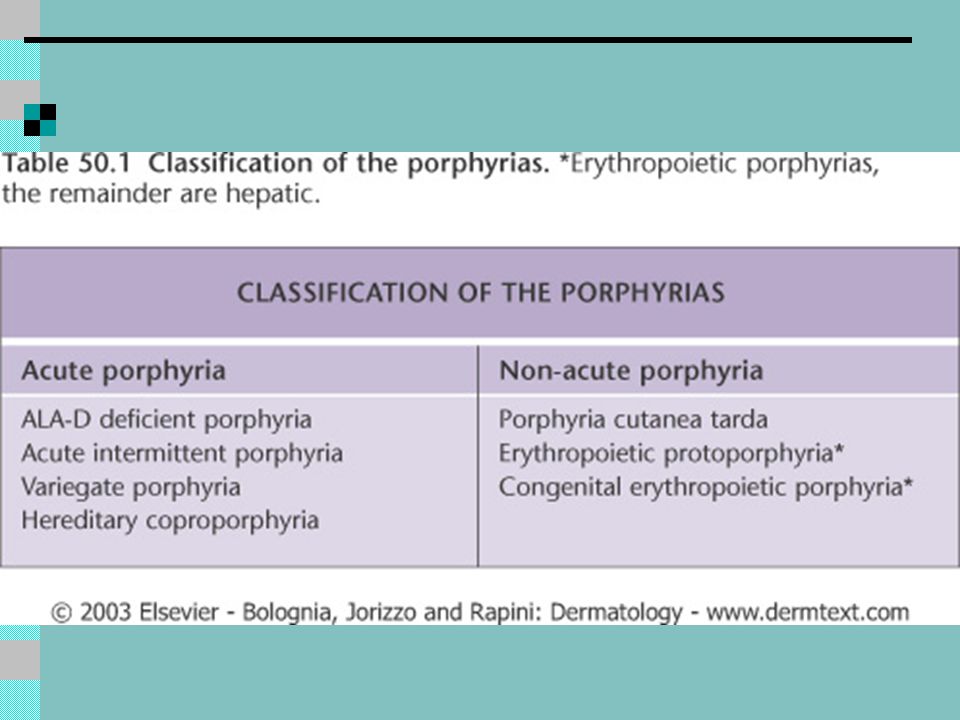
Erythropoietic forms Congenital erythropoietic porphyria (CEP) Erythropoietic protoporphyria (EPP) Erythropoietic coproporphyria ECP Hepatic forms Acute intermittent porphyria (AIP) ALA dehydrogenase deficiency Hereditary coproporphyria (HCP) Variegate porphyria (VP) Porphyria cutanea tarda
 Erythropoietic protoporphyria (EPP) Erythropoietic coproporphyria ECP. Hepatic forms. Acute intermittent porphyria (AIP) ALA dehydrogenase deficiency. Hereditary coproporphyria (HCP) Variegate porphyria (VP) Porphyria cutanea tarda.")
79
HEP, hepatoerythrocytic porphyria, has been classified as either a hepatic or hepatoerythropoietic type Diagnosis should be made by identifying characteristic clinical and biochemical abnormalities

80
Porphyria cutanea tarda
The most common type of porphyria Characterized by photosensitivity resulting in bullae These rupture easily to form erosions or shallow ulcers and heal with scarring, milia and dyspigmentation Patients may complain of skin fragility Hypertrichosis and sclerodermatous thickening

81
Liver disease is frequent
Alcoholism is common, Hep C in 94% in US Frequently associated with other diseases DM in 15-20%, lupus erythematosis HIV Estrogen treatment is associated with the appearance of PCT by an unknown mechanism

82
PCT in a patient with Hepatitis C virus infection
PCT in a patient with Hepatitis C virus infection. Multiple erosions with hemorrhagic crusts are seen as well as an intact blister on the lateral fourth finger

83
PCT in chronic renal failure

84
PCT

85
Deficiency in uroporphyrinogen decarboxylase
Most common is the sporadic nonfamilial form, 80%, abnormal enzyme activity in the liver Patients present in midlife, 45 Familial type, AD, deficiency in liver and RBCs Nonfamilial, (acquired toxic PCT), associated with acute or chronic exposure to hepatotoxins
, associated with acute or chronic exposure to hepatotoxins.")
86
Diagnosis is strongly suspected on clinical grounds
Coral red fluorescence in random urine 24 hour urine, 300 to several thousand micrograms Uroporphyrins to coproporphyrins 3:1 to 5:1 Biopsy – noninflammatory, subepidermal bulla with an indulating, festooned base Caterpillar bodies, basement membrane material DIF of involved skin shows IgG and C3 at the DEJ, and in the vessel walls in a linear pattern

87
Histologic features of PCT
Subepidermal blister with minimal dermal inflammatory infiltrate. Festooning of dermal papillae

88
treatment Remove all precipitating environmental factors
Physical barrier protection, barrier sunscreens Phlebotomy, uroporphyrinogen decarboxylase is inhibited by iron 500 ml at 2 week intervals, hemoglobin 10 g/dL Several months, 6-10 phlebotomies Antimalarials, full doses may produce severe hepatotoxic reaction

89
Remission may last years, with relapse repeat treatment
Iron chelation May respond to transplant in renal failure May improve with treatment if assoc. with Hep C

90
pseudoporphyria Blistering and skin fragility identical to PCT, histologic features of PCT Normal urine and serum porphyrins Hypertrichosis, dyspigmentation, and cutaneous sclerosis do not occur Most commonly caused by medications Typically NSAIDs, noproxen Sunbed use, hemodialysis

91
treatment Physical sun protection Discontinue inciting medication
May resolve over several months

92
Hepatoerythropoietic porphyria
Very rare form Inherited as an AR trait Deficiency of uroporphyrinogen decarboxylase, 10% of normal in both the liver and erythrocytes Dark urine at birth Vesicles, sclerodermoid scarring, hypertrichosis, pigmentation, red fluorescence of teeth

93
Abnormal urinary porphyrins as in PCT
Elevated erythrocyte protoporphyrins Increased coproporphyrins

94
Hepatoerythropoietic porphyria

95
Acute intermittent porphyria
Second most common form Characterized by periodic attacks of abdominal colic, gastrointestinal disturbances, paralyses, and psychiatric disorders No skin lesions are seen AD trait, deficiency in porphobilinogen deaminase Only 10 % develop disease, all are at risk for primary liver cancer

96
Severe abdominal colic is most often the initial symptom of AIP, NVDC may accompany the pain
Elevated levels of urinary porphobilinogen Increased dALA in plasma and urine No specific treatment is available Avoid precipitating factors Glucose loading

97
Hematin infusions Pain management Oral contraceptives may prevent attacks in women with premenstrual symptoms

98
Hereditary coproporphyria HCP
Rare, AD Deficiency of coproporphyrinogen oxidase One third are photosensitive Prone to GI attacks similar to AIP and VP Fecal coproporphyrin is always increased Urinary coproporphyrin, ALA, and PBG are only increased during attacks

99
Variegate porphyria VP
Mixed porphyria, South African genetic porphyria, mixed hepatic porphyria, AD inheritance Decreased activity of protoporphyrinogen oxidase Majority of relatives have silent VP, reduced enzyme activity but no clinical lesions

100
Characterized by the combination of the skin lesions of PCT and the GI and neurologic disease of AIP
705 of patients develop skin lesions Vesicles and bulla with erosions Hypertrichosis and hyperpigmentation Facial scarring and thickening of skin

101
Suspect VP when finding indicate both PCT and AIP, esp
Suspect VP when finding indicate both PCT and AIP, esp. with history of South African ancestry Fecal coproporphyrins and protoporphyrins are always elevated During attacks, urine porphobilinogen and ALA are elevated Urinary coproporphyrins are increased over uroporphyrins

102
A finding in the plasma of “X porphyrin”, fluorescence at 626 nm, is characteristic and distinguishes this form from others Symptomatic treatment as is for PCT and AIP

103
Erythropoietic protoporphyria EEP
AD and AR forms Ferochelatase activity is 10 to 25% of normal in affected persons Typically presents early in childhood, 2-5 years Immediate burning of the skin upon sun exposure Elevated protoporphyrin IX absorbs both the Soret band and also at nm

104
Infants cry when exposed to sunlight
Severe liver disease develops in 10% Excessive porphyrins are deposited in the liver Suspect diagnosis on clinical grounds, with acute symptoms and chronic skin changes Urine porphyrin levels are normal Erythrocyte protoporphyrin is elevated Erythrocyte, plasma, and fecal protoporphyrin can be assayed to confirm the diagnosis

105
Skin biopsy will confirm diagnosis
Treatment, protection with barrier sunscreens Beta carotene, phototherapy, cysteine Transfusions for anemia

106
Erythropoietic protoporphyria
Subtle scarring

107
Erythropoietic protoporphyria
Erythema and hemorrhagic crusts

108
Congenital erythropoietic porphyria, CEP
Gunther’s disease AR, defect of the enzyme uroporphyrinogen III synthase Presents soon after birth with the appearance of red urine Severe photosensitivity Blistering, scarring, ectropion and corneal damage may occur Erythrodontia is characteristic

109
Mutilating scars, hypertrichosis, profuse eyebrows, long eyelashes, “monkey face” or “werewolf”
Other features – growth retardation, hemolytic anemia, thrombocytopenia, porphyrin gallstones, osteopenia Diagnosis of CEP should be suspected with an infant with dark urine and severe photosensitivity

110
Congenital erythropoietic porphyria
Erythrodontia Severe mutilation

111
Fluorescence of circulating red blood cells, CEP with UVA
Vs. transient fluorescence in EPP

112
High amounts of uroporphyrin I and coproporphyrin I are found in the urine, stool and red cells
Treatment – strict avoidance of sunlight and sometimes splenectomy for the hemolytic anemia Oral activated charcoal Repeated transfusions to maintain hematocrit level at 33% turns off demand for heme Bone marrow transplantation

113
Transient erythroporphyria of infancy (purpuric phototherapy-induced eruption)
Report of seven infants exposed to 380 to 700 nm blue lights for the treatment of indirect hyperbilirubinemia who developed marked purpura on the exposed skin All infants had received transfusions Elevated plasma coproporphyrins and protoporphyrins were found in 4 Pathogenesis is unknown

114
END

Similar presentations