Download presentation
Presentation is loading. Please wait.

1
Putting genetic interactions in context through a global modular decomposition Jamal

2
Genetic interaction provide powerful perspective how gene functions specific mechanisms that give rise to these interactions not well understood Requires a thorough study of genetic interaction networks understand the structure of the network. Motivation

3
This study This study uses a datamining approach to explore all block structure with in this network.

4
Characteristics Genetic interaction: “ Multiple genetic perturbations whose combination result in a phenotype that is unexpected given the phenotypes of the individual perturbations” The redundancies and dependencies within genetic network can provide powerful means for functional characterization.

5
Unlike the PPI network, there is no obvious functional interpretation of a single genetic interaction, either negative or positive. The genetic interaction of two genes does not imply that they interact physically, it simply suggest that they share some kind of functional interaction.

6
Modular hypothesis Gene membership falls into different type of functional modules For example: Protein complexes, pathways, etc.

7
Negative between pathway Model Defines Negative interactions: which are thought to arise between functionally redundant pathways such that deleting any pair of genes spanning across the pathways results in a significant reduction of fitness

8
Positive within pathway Model defines Positive interactions: If the second deletion in that same compromised pathway does not result in any additional fitness defect.

9
Bi-Clusters as block pattern in network Can be over-lapping or disjoint sets of genes Every gene in one set is connected to every other gene in other set.

10
Pu et al.(2008) specifically designed an algorithm that randomly start with an initial bi-cluster and then rediscover the prominent bi-cluster many times. In this study authors employed an approach based on an algorithm from field association rule mining to find all biclusters of sufficient size.

11
Approach Summary--bi-cluster Discovery Recent data from Costanzo et al. (2010) was used in this study and the developed approach utilizes the apriori algorithm from the field of association rule mining to discover all biclusters. and the biclusters that can be expressed by degree distribution alone were filter out using non-parametric statistical assessment.
 was used in this study and the developed approach utilizes the apriori algorithm from the field of association rule mining to discover all biclusters. and the biclusters that can be expressed by degree distribution alone were filter out using non-parametric statistical assessment..")
12
XMOD This approach XMOD (eXhaustive Modular Discovery) guaranteed to find all bi-partite graphs : Where 1 part of bi-partite acts as a functional unit
 guaranteed to find all bi-partite graphs : Where 1 part of bi-partite acts as a functional unit")
13
Presence of degree distribution based Bi-clusters Edges were randomized and still bi-partite graphs were obtained suggesting that biologically meaningless bipartite graphs can exist. score for each bi-cluster lower for biologically meaningful Score: “ the product of probabilities of each edge occurring independently conditioned on the degree of two interacting genes”

14
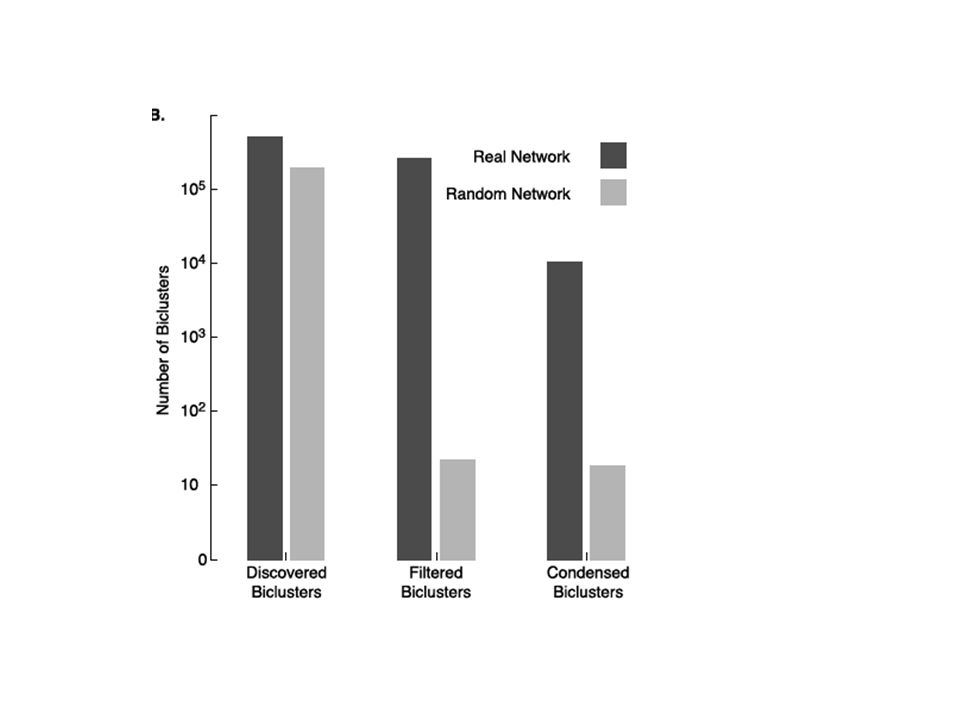
Filtered Biclusters: using the independence score a cutt off is applied to separate the ones with less independence score Condensed Biclusters: after removing Biclusters with >40% overlap

16
Comparison with other techniques

17
Dataset The dataset in Costanzoo et al. in 2010 was used. 85,714 negative interactions and 35,858 interactions were used.

18
Association rule Mining Apriori Algorithm in Agrawal (1993) was used. Its standard available implementation from a website was used. Apriori was run on a binary set of positive interactions and also on a set of negative interactions

19
Randomizing the Genetic Interaction network The number of edges for each gene was preserved but the targets were randomized. A gene cannot have an edge with itself

20
Filtering Random bi-clusters We found that 50% of the real negative biclusters and 6% of real positive biclusters have scores below the 0.01 percentile of biclusters of the same size from the random networks. This resulted in 256,502 negative biclusters and 2194 positive biclusters.

21
Removing overlap from Biclusters we first arranged the biclusters in descending order by area. Then, beginning with the first bicluster A, we removed all biclusters whose area overlap with A was greater than 0.4, where overlap between biclusters A and B was calculated using the following formula:

22
Evaluation of Functional Coherence MEFIT network is based on coexpression data and does not use genetic interaction datasets

23
Improvements?

Similar presentations










 11/05/07. Methods Linear –PCA (Raychaudhuri et al. 2000) –NIR (Gardner et al. 2003) Nonlinear –Bayesian network (Friedman.>")


 10/07/09. Outline Affinity propagation Quality evaluation.>")





