Download presentation
Presentation is loading. Please wait.

1
Bone

2
Bone Organic matrix( 35%)- cells & proteinaceous osteoid
Inorganic elements( 65%)-calcium hydroxyapatite, 99% of body’s ca- 85% of P- 65% of Na and Mg Remodeling: constant breakdown & renewal which the net effects may be bone maintenance, bone loss or bone deposition
-calcium hydroxyapatite, 99% of body’s ca- 85% of P- 65% of Na and Mg. Remodeling: constant breakdown & renewal which the net effects may be bone maintenance, bone loss or bone deposition.")
3
Bone diseases Congenital diseases Acquired diseases Fractures
Osteonecrosis Osteomyelitis Tumors

4
Congenital diseases of bone

5
Developmental anomalies
localized problems in migration of mesenchymal cells & formation of condensations, dysostoses Isolated sporadic lesion or a component of a complex syndrome More common lesions Aplasia- congenital absence of a digit or rib Formation of extra bones- supernumerary digits or ribs Abnormal fusion of bones- premature closure of cranial sutures or congenital fusion of ribs

6
Mutations Interfere with bone or cartilage formation, growth, and/or maintenance of normal matrix components More diffuse defects Dysplasia osteodysplasia, chondrodysplasia

7
Other genetic metabolic disorders
Not usually thought of as primary skeletal diseases, eg; mucopolysaccharidoses like Hurler syn

8
Osteogenesis imperfecta (brittle bone disease)
")
9
Osteogenesis imperfecta (brittle bone disease)
A group of hereditary disorders caused by defective synthesis of type I collagen Gene mutations in coding sequences for α1 or α2 chains, quality or quantity( premature degradation, dominant negative mutation ) Most, AD Extraskeletal manifestations: skin, joints, eyes….
 Most, AD. Extraskeletal manifestations: skin, joints, eyes….")
10
OI Too little bone, extreme skeletal fragility
Four major subtypes, extremely broad range of clinical outcome Type I- normal lifespan, fractures during childhood, blue sclera, hearing loss, small misshapen teeth Type II-fatal

11
Achondroplasia

12
Achondroplasia Activating Point mutation in FGF receptor3
Activation of receptor Inhibits chondrocyte proliferation Impaired long bone growth

13
Achondroplasia AD Spontaneous mutation( many cases )
Affected individuals are typically heterozygotes Homozygotes die soon after birth because of abnormalities in chest development & respiratory failure

14
Clinical findings Most common form of dwarfism
Affects all bones that form from a cartilaginous framework Most conspicuous changes: marked disporportionate shortening of proximal extremities, bowing of the legs, lordotic posture Cartilage growth plate: disorganized & hypoplastic

15
Osteopetrosis

16
Osteopetrosis A group of rare genetic disorders characterized by reduced osteoclast-mediated bone resorption, defective bone remodeling Several variants, most common: 1- AD adult form with mild clinical manifestations 2- AR infantile with a severe/ lethal phenotype

17
Osteopetrosis Causing defects Those that disturb osteoclast function
Those that interfere with osteoclast formation & differentiation

18
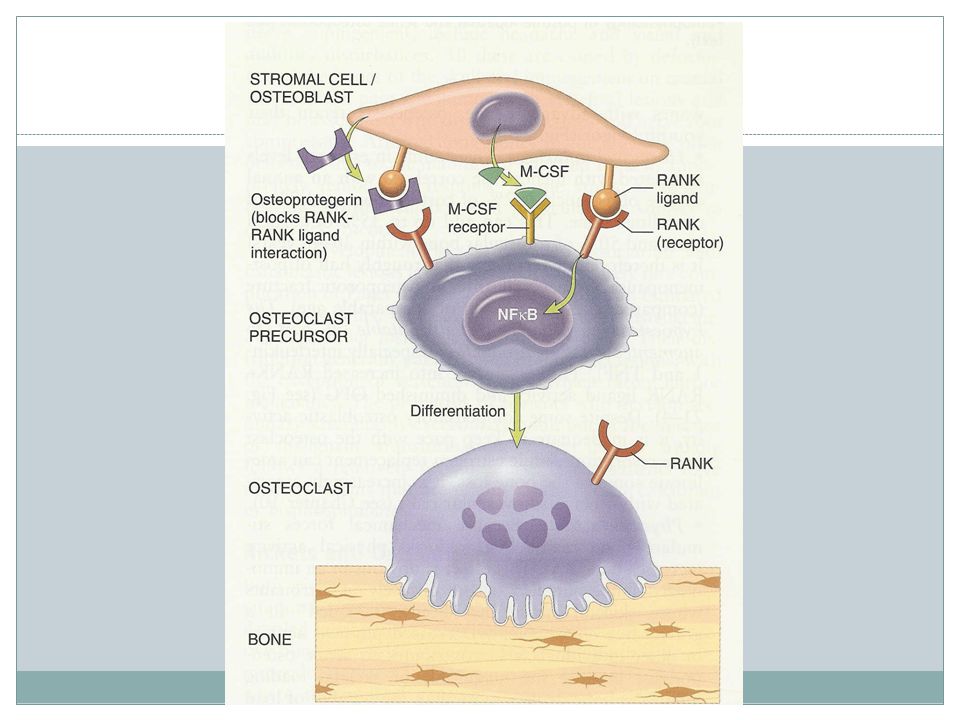
osteoclast dysfunction
Bone resorption through osteocalsts: decalcification by proton pump and degrading enzymes also activation of mediators Nature of osteoclast dysfunction unknown in many cases Carbonic anhydrase II deficiency results in reduced bone demineralization( required for osteoclast H+ excretion) Proton pump deficiency Chloride channel defect
 Proton pump deficiency. Chloride channel defect.")
19
Clinical findings Fractures Cranial nerve problems
Recurrent infections( diminished hematopoiesis ) Hepatosplenomegaly Bone marrow transplant
 Hepatosplenomegaly. Bone marrow transplant.")
20
Acquired diseases of bone development
Nutritional deficiencies( vit c, vit d) Primary & secondary hyperparathyroidism Osteoporesis Paget disease Rickets & osteomalacia
 Primary & secondary hyperparathyroidism. Osteoporesis. Paget disease. Rickets & osteomalacia.")
21
Osteoporosis Increased porosity of skeleton resulting from reduced bone mass, increase in bone fragility & fx Localized to a bone or region or generalized Most common forms: senile, postmenopausal Bone loss generally occurs in areas containing abundant cancelloues bone so more pronounced in spine & femoral neck

28
Paget disease ( osteitis deformans)
")
29
Paget disease Gain in bone mass but newly formed bone is disordered & lacks strength Repetitive episodes of regional osteoclastic activity & bone resorption- followed by exuberant bone formation- finally by exhaustion of cellular activity Osteolytic stage, mixed osteoclastic- osteoblastic stage, osteosclerotic stage Age: mid adulthood Marked variation in prevalence in different populations

30
Morphology Lytic phase- numerous & large osteoclasts
Mixed phase- prominent osteoblasts, marrow replaced by loose connective tissue Mosaic pattern( pathogonomic histologic feature )
")
31
Pathogenesis Paramyxovirus infection IL-1 secretion from infected cells, M-CSF activate osteoclasts Other suggested mechanism: intrinsic hyperresponsiveness of osteoclasts to activating agents as, vitD & RANK ligand.

32
Clinical course Monostotic 15% ( tibia, ilium, femur, skull, vertebra, humerus ) Polyostotic ( pelvis, spine, skull ) axial skeleton or proximal femur , 80% of cases Ribs, fibula & small bones of hands & feet : unusual Most cases are mild & discovered incidentally Elevation in serum ALKP & increased urinary excretion of hydroxyproline
 axial skeleton or proximal femur , 80% of cases. Ribs, fibula & small bones of hands & feet : unusual. Most cases are mild & discovered incidentally. Elevation in serum ALKP & increased urinary excretion of hydroxyproline.")
33
Manifestations Warmth of overlying skin & subcutis
In extensive polyostotic disease high output congestive heart failure In proliferative phase of skull disease, nerve impigment headache & visual and auditory disturbances Back pain with vertebral lesions, fx & nerve root compression Deformity of long bones of leg Sarcoma in 1% of patients parallel to lesions except vertebra

35
Rickets & Osteomalacia
Defective bone mineralization

36
Hyperparathyroidism

37
PTH Osteoclast activation( increased RANKL production by osteoblasts )
Increased resorption of ca by renal tubules Increased urinary excretion of phosphate Increased synthesis of 1,25(OH)2 vitD by kidneys Net result: elevation in serum ca, inhibiting PTH
2 vitD by kidneys. Net result: elevation in serum ca, inhibiting PTH.")
38
Hyperparathyroidism Significant skeletal changes related to unabated osteoclast activity Entire skeleton is affected, some sites may be more severely affected PTH is directly responsible for bone changes in primary but additional influences contribute in secondary Inadequate 1,25(OH)2 vitD synthesis in chronic renal failure
2 vitD synthesis in chronic renal failure.")
39
Hallmark: Increased osteoclastic activity & bone resorption

40
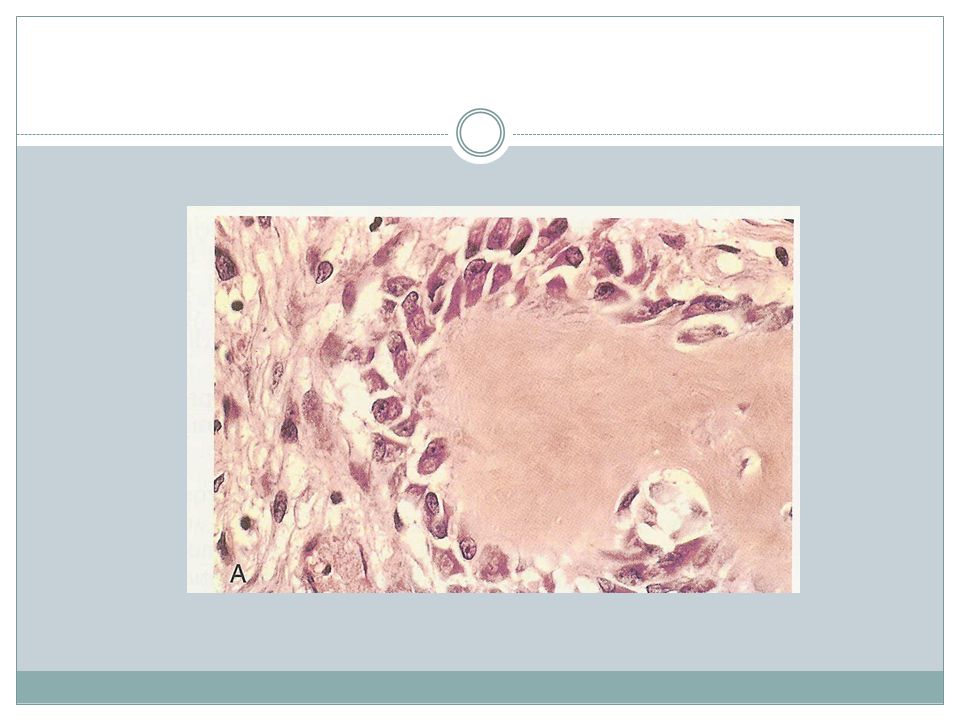
Brown tumor Osteitis fibrosa cystica

41
Fractures

42
Healing Blood coagulum
recruit inflammatory cells, fibroblast & endothelium Release of cytokines from plts & inflammatory cells Activate bone progenitor cells Soft tissue callus, within a week Deposition of woven bone Chondroblasts Early repair process peak within 2-3 wks Bony callus Weight bearing leads to resorption of callus from nonstressed sites

43
Disrupting factors Displaced & comminuted fractures
Inadequate immobilization nl constiuents do not form Too much motion along fx gap Infection Inadequate levels of ca or p, vit deficiencies, systemic infection, diabetes, vascular insufficiency

44
Osteonecrosis ( avascular necrosis)
")
45
Mechanisms Vascular compression or disruption Steroid administration
Thromboembolic disease Primary vessel disease (eg; vasculitis )
")
46
Osteonecrosis Cortex, usually not affected Subchondral infarcts
Medullary infarcts

47
Osteomyelitis Pyogenic tuberculous

48
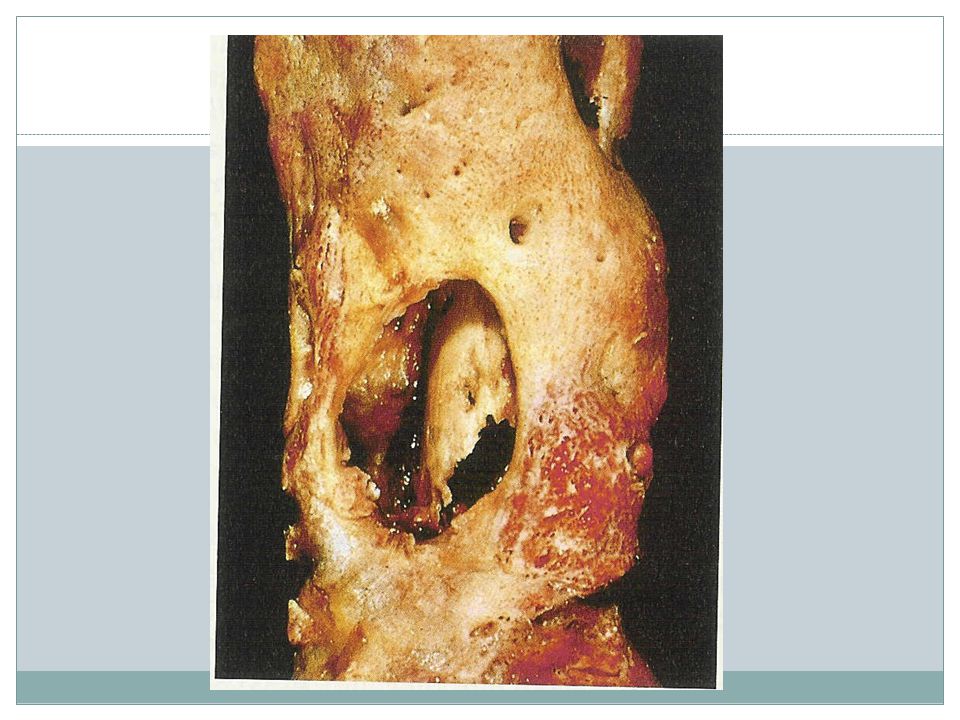
Pyogenic osteomyelitis
Routes Organisms: staph aureus, E-coli and strep group B, salmonella, mixed bacterial infections No organism isolated, 50% of cases Associated suppurative arthritis in infants Sequestrum Involucrum Subperiosteal abscess and draining sinus After the 1st week of infection chronic inflammatory cell become numerous ¼ of cases do not resolve and persist as chronic infection

50
Complications of chronic OM
Acute flare ups Pathologic fx Secondary amyloidosis Endocarditis Sepsis SCC Osteosarcoma, rarely

51
Tuberculous OM 1-3% of pulmonary infections
Usually reach the bone through blood stream( long bones & vertebra) , although direct spread may be Solitary Pott disease, vertebral deformity & collapse with secondary neurologic deficit, soft tissue abscess( psoas muscle ), common
 , although direct spread may be. Solitary. Pott disease, vertebral deformity & collapse with secondary neurologic deficit, soft tissue abscess( psoas muscle ), common.")
52
End of first session

53
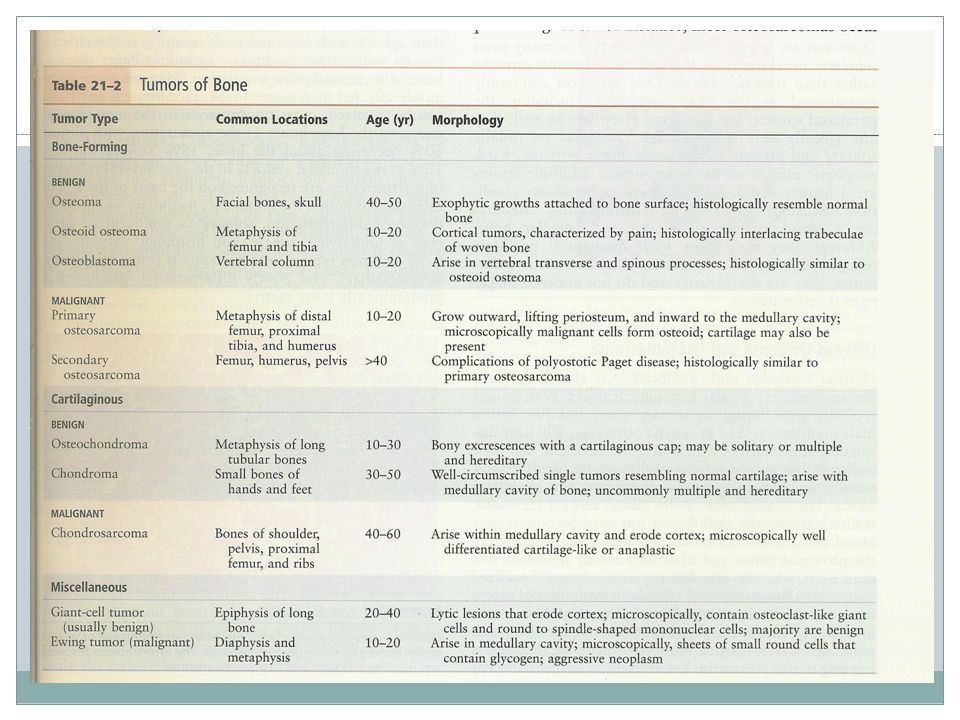
Bone tumors Primary metastatic

54
General considerations
Classification: cell of origin and apparent pattern of differentation Osteosarcoma: the most common primary bone cancer then CSA and EWS Osteochondroma & fibrous cortical defect: frequent Most bone tumors occur during first several decades and have a propensity to originate in long bones of extremities Specific tumor types target certain age groups & anatomic sites, OSA , CSA

55
General considerations
Most bone tumors arise without any prior known cause but Genetic syndromes( Li-Fraumeni & retinoblastoma syndromes ), bone infarcts, chronic osteomyelitis, paget dis, radiation and metal orthopedic devices are associated with OSA Clinical presentations
, bone infarcts, chronic osteomyelitis, paget dis, radiation and metal orthopedic devices are associated with OSA. Clinical presentations.")
57
Major tumor types Abnormal development Benign neoplasm
Malignant neoplasm Neoplasms of uncertain potential

58
Bone- forming tumors Osteoma Osteoid osteoma osteosarcoma

59
Osteoma Many cases are developmental aberrations or reactive growths rather than true neoplasms Most common in head & neck including paranasal sinuses Middle age Solitary Localized, slowly growing hard exophytic masses on bone surface Multiple lesions are a feature of Gardner syn

60
Osteoid osteoma & Osteoblastoma
Age: teenage & 20s Male predilection Distinguished by size, site of origin, radiographic appearance

61
Clinical presentation
Site Size Tumor Localized pain Responsive to aspirin Proximal femur & tibia Less than 2 cm Osteoid osteoma Pain ,Not responsive to aspirin Vertebral column larger Osteoblastoma

63
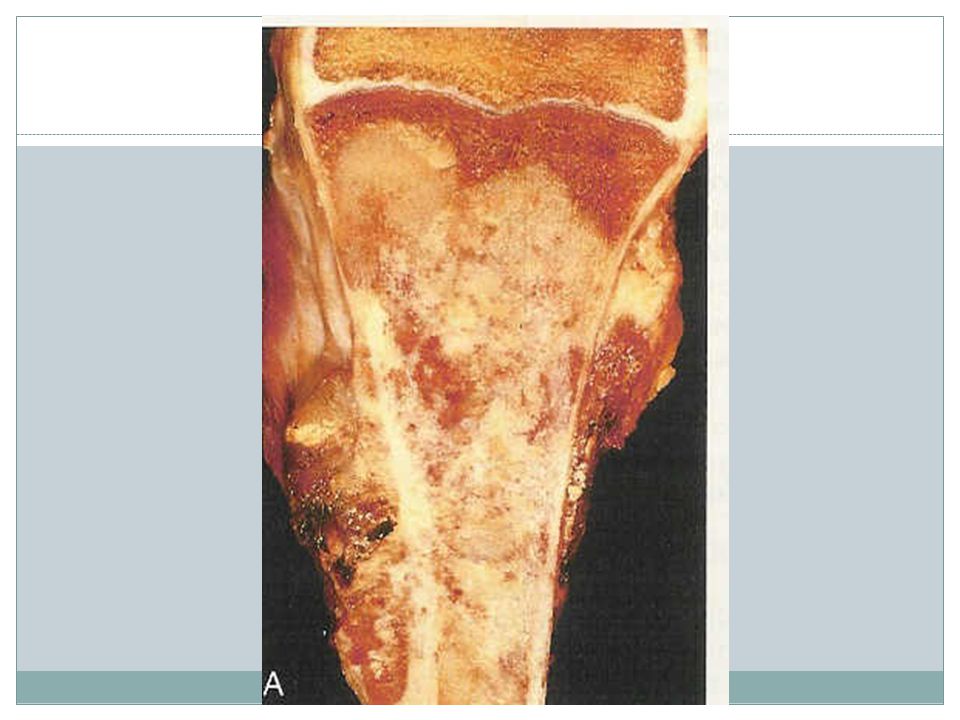
Osteosarcoma Age- 75% younger than 20, a second peak in elderly usually with other conditions including Paget dis, bone infarct, prior irradiation Male>female Location: most tumors arise in metaphysis of long bones of extremities 60% around the knee, 15% around the hip, 10% at shoulder, 8% jaw Subtypes- the most common type: primary, solitary, intramedullary and poorly differentiated

66
Pathogenesis RB gene mutations occur in 60-70% of sporadic tumors
Patients with hereditary retinoblastomas have a thousandfold greater risk of developing OSA Many OSA develop at sites of greatest bone growth

67
Clinical features Typical presentation: painful enlarging mass X-ray
Hematogenous spread, 10-20% of patients have demonstrable pulmonary metastasis at time of DX Long-term survival: 60-70% Secondary OSA, highly aggressive that do not respond well to therapy

68
Cartilage forming tumors
Osteochondroma Chondroma chondrosarcoma

69
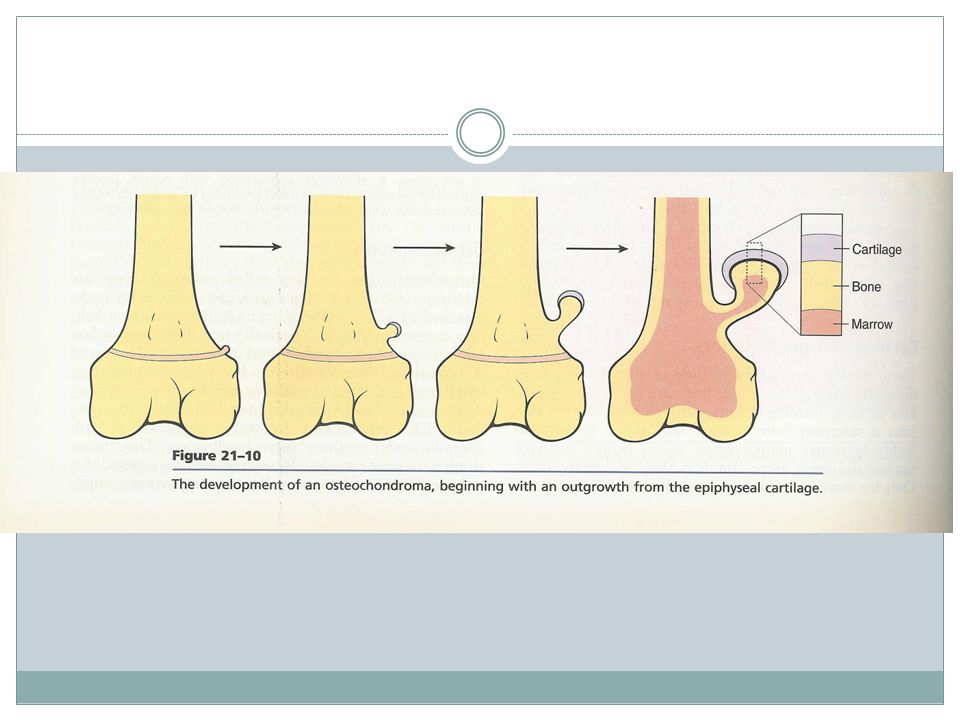
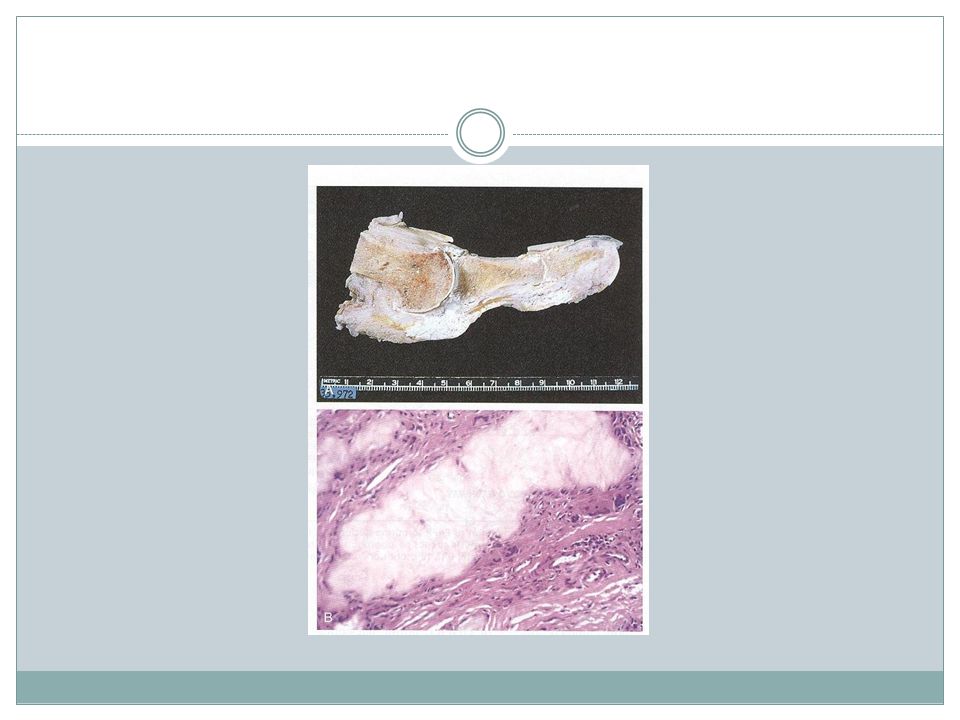
Osteochondroma( Exostose )
Age- late adolescent & early adulthood but multiple become apparent during childhood Inactivation of both copies of EXT gene in chondrocytes is implicated in both sporadic and hereditary EXT gene- a tumor suppressor gene encoding glycosyltransferases essential for polymerization of heparin sulfate

70
Osteochondroma Location- bones of endochondral origin arising at metaphysis near the growth plate of long tubular bones Occasionally develop from bones of pelvis, scapula and ribs( sessile ) Short tubular bones of hands and feet: rare Clinical presentation
 Short tubular bones of hands and feet: rare. Clinical presentation.")
72
Chondroma Enchondroma, juxtacortical chondroma Age- 20-50 yrs old
Location- solitary at metaphysis of tubular bones esp short tubular bones Ollier disease -multiple chondromas preferentially involving one side of the body Maffucci syndrome- multiple chondromas and benign soft tissue angiomas

73
Clinical features Incidental findings X-ray Malignant transformation

74
Chondrosarcoma Age- 40 or older Male>female
Subclassification: intramedullary, juxtacortical Variants: conventional, myxoid, dedifferentiated, clear-cell, mesenchymal. Dediferentiation occur in about 10% of low-grade CSA

75
Clinical features Site- pelvis, shoulder and ribs X-ray
Direct correlation between grade and biologic behavior 5-year survival-Low-grade tumors: 80-90% grade 3 tumors: 43% Metastasis in grade 1 tumors:rare grade 3 tumors: 70% Size >10cm Hematogenous spread, lung, skeleton

78
Fibrous and fibro-osseous tumors
Fibrous cortical defect & Nonossifying fibroma Fibrous dysplasia

79
Fibrous cortical defect
30-50% of all children older than 2 yrs old Developmental defects rather than true neoplasm Mostly smaller<0.5cm and arise in metaphysis of distal femur or proximal tibia Bilaterality or multiplicity: 50% Asymptomatic and usually incidental findings Most undergo spontaneous differentiation

81
Fibrous dysplasia Localized developmental arrest Clinical patterns:
Monostotic Polyostotic Polyostotic disease associated by café au lait skin pigmentation and endocrine abnormalities esp precocious puberty( McCune-Albright syn ) Rarely polyostotic disease can transform into osteosarcoma.
 Rarely polyostotic disease can transform into osteosarcoma.")
82
Monostotic FD 70% of cases Age- early adolescence
Most common sites: ribs, femur, tibia, jaw bones, calvaria & humerus Asymptomatic & usually incidental findings Can cause marked enlargement and distortion of bone

83
Polyostotic FD Majority of remaining cases Age- slighly earlier
Site- femur, skull, tibia, humerus Craniofacial involvement- 50%, 100%

84
McCune-Albright syn 3% of cases
Sexual precocity, hyperthyroidism, GH secreting pituitary adenoma, primary adrenal hyperplasia. The severity of manifestations depends on the number and cell types that harbor G-protein mutation Bone lesions, often unilateral & skin lesions usually limited to the same side of the body. Macules are classically large, dark to light brown and irregular.

86
Miscellaneous bone tumors
Ewing sarcoma Primitive neuroectodermal tumor Giant cell tumor of bone Metastatic disease

87
Ewing Sarcoma and Primitive Neuroectodermal tumor
PNET- neural differentiation EWS- undifferentiated Age- most yrs %<20yrs Translocation-95% of patients have t( 11;22 ) (q24;q12 ) or t(21;22)(q22;q12) A chimeric protein which is an active transcription factor
 (q24;q12 ) or t(21;22)(q22;q12) A chimeric protein which is an active transcription factor.")
88
Clinical features Typical presentation: painful enlarging mass in diaphysis of long tubular bones( esp femur ) & pelvic flat bones Systemic signs and symptoms in some X-ray- onion skin pattern of periosteal reaction 5-yrs survival: 75%

90
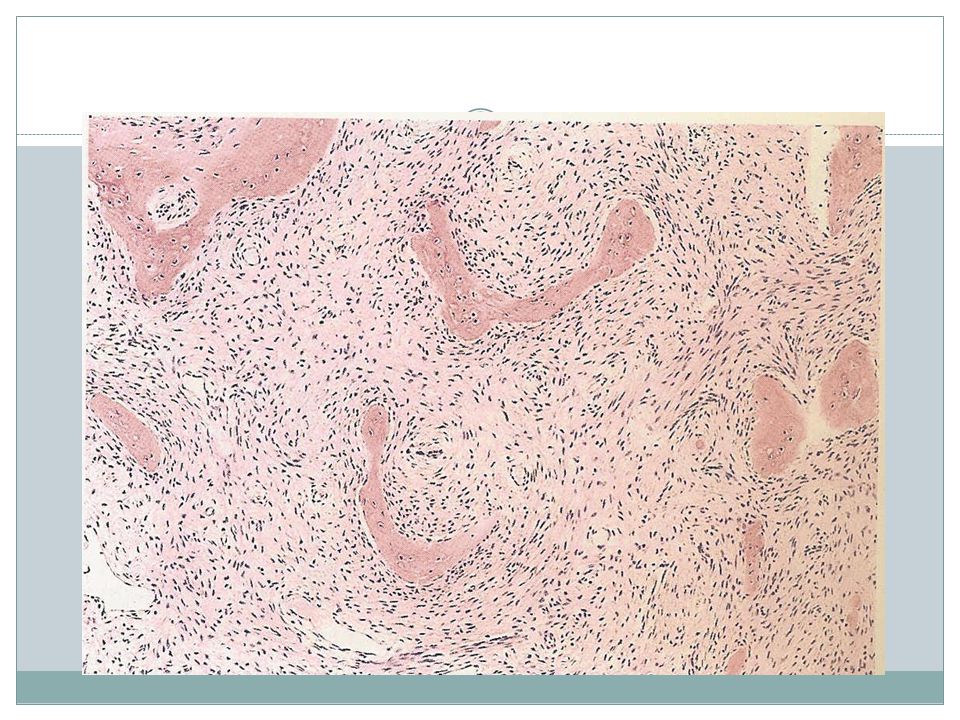
Giant-Cell Tumor of Bone
Benign, locally aggressive Age yrs Location- epiphysis of long bones around the knee X-ray- large purely lytic and eccentric lesion Cortical destruction± Recurrence: ½ of cases Metastasis to lungs: 4%

92
Metastatic disease Pathways of spread -Direct extension
-Lymphatic or hematogenous -Intraspinal Origin Adults- prostate, breast, kidney, lung Children- Neuroblastoma, Wilm’s tumor, OSA, EWS, RMS

93
Metastasis site axial skeleton proximal femur humerus X-ray pure lytic
pure blastic both

94
END OF SECOND SESSION

95
DISEASES OF THE JOINTS Basic pathology

96
Degenerative joint disease
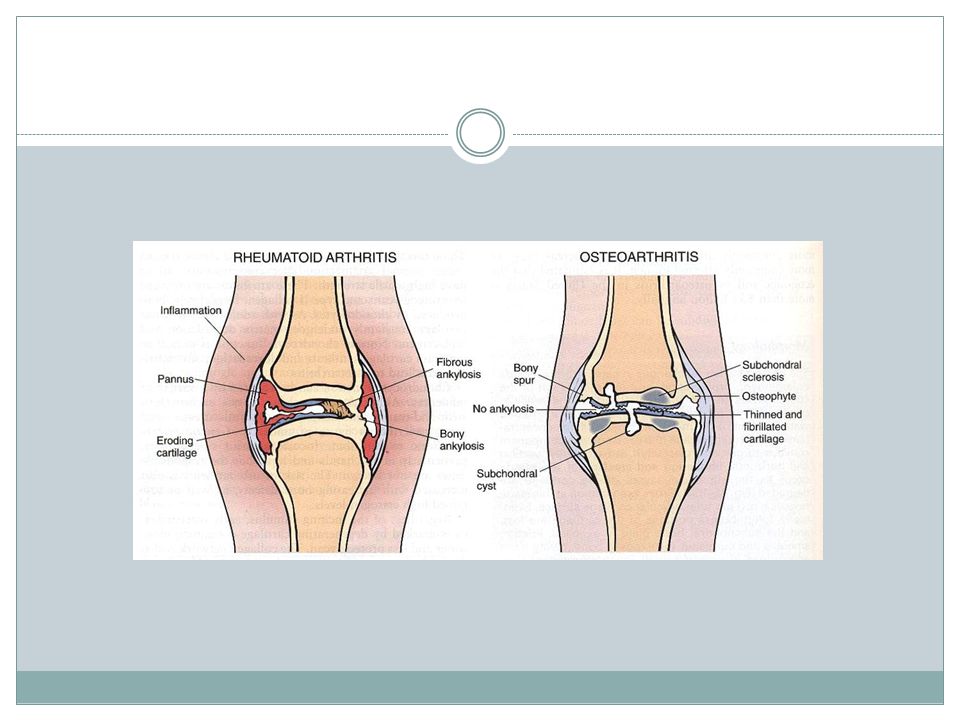
The most common type of joint disease . Progressive erosion of articular cartilage. Important cause of physical disability in olders. Suffix itis is misleading. Cartilage biochemical & metabolic changes result in it’s breakdown.

97
D.J.D Primary:appears without apparent initiating cause as an aging process Secondary:appears in younger individuals having some predisposing conditions eg,trauma,underlying systemic disease,diabetes………

98
pathogenesis (↓collagen synthesis,↑collagen degradation)
Aging prevalence increases > 50 Mechanical stresses Genetic factors Chondrocytes play a primary role (↓collagen synthesis,↑collagen degradation) probably ↑apoptosis
 probably ↑apoptosis.")
99
Gross Granular articular surface,softer than nl Loss of cartilage
Ivory appearance of exposed bone Subchondral cyst Dislodged pieces of cartilage & bone into the joint(joint mice)
")
100
D.J.D

101
Microscopy Fibrilation & cracking of the matrix
Sloughing of full thickness of cartilage Bone eburnation Small fractures through the articulating bone Joint mice(dislodged pieces of bone & cartilage) Osteophytes Minor changes in synovium as congestion ,fibrosis & scattered chronic inflammatory cells
 Osteophytes. Minor changes in synovium as congestion ,fibrosis & scattered chronic inflammatory cells.")
102
Articular cartilage Fibrilation of the surface
Fissures (horizontal & vertical)
")
103
Cartilage cloning Duplication of tidemark

104
Vascular penetration at base of cartilage
Cysts in the subchondral bone Nonspecific synovitis

105
Clinical course Deep achy pain,worsens with use.
Involvement of one or a few joints Commonly:hip,knee,lower lumbar & cervical vertebra,interphalangeal joints of finger(proximal & distal), first carpometacarpal & tarsometatarsal joints
, first. carpometacarpal & tarsometatarsal. joints.")
106
Rheumatoid arthritis A Chronic systemic inflammatory disorder
May affect many tissues & organs such as:skin,blood vessels,heart,lungs & muscle. principally attack the joints. Nonsuppurative proliferative synovitis that often progress to destruction of cartilage & joint ankylosis Symmetric polyarticular arthritis Most of them Chronic relapsing & remitting course & eventually leads to severe joint destruction

107
Pathogenesis Genetic predisposition Strong association of HLA-DR1&DR4
Environmental factors ??? EBV,Borrelia,mycoplasma,parvovirus.? Autoimmune reaction ALTHOUGH INITIATING AGENT IS STILL UNKNOWN

108
Morphology in the joints
Dense perivascular inflammatory infiltration as:lymphoid follicles,plasma cells& macrophages in the synovial stroma Increased vascularity(vasodilation & angiogenesis) Fibrin aggregates in synovium & floating in joint space as rice bodies Osteoclast activity in underlying bone Pannus formation
 Fibrin aggregates in synovium & floating in joint space as rice bodies. Osteoclast activity in underlying bone. Pannus formation.")
109
Synovium Protrusion of large & edematous villi into the joint
Variable color: yellowish , gray or brown

110
Infiltration of mononuclear inflammatory cells
Vascular proliferation

111
Nodular lymphocytosis with germinal centers
Plasma cell cuffing

112
Hypertrophy & hyperplasia of synovial cells
Synovial giant cells(Grimley-sokoloff GC)
")
113
Fibrin exudate as loose bodies (rice bodies) or attached by inflammatory stalk to synovium
 or attached by inflammatory stalk to synovium")
114
Pannus A neoplasm-like growth of inflamed synovial tissue
leads to destruction of joint structures Two types : - vascular inflammatory type - avascular fibrous type

115
pannus

116
Eventually Fibrous obliteration of the joint
Deformed joints with minimal or no range of motion

118
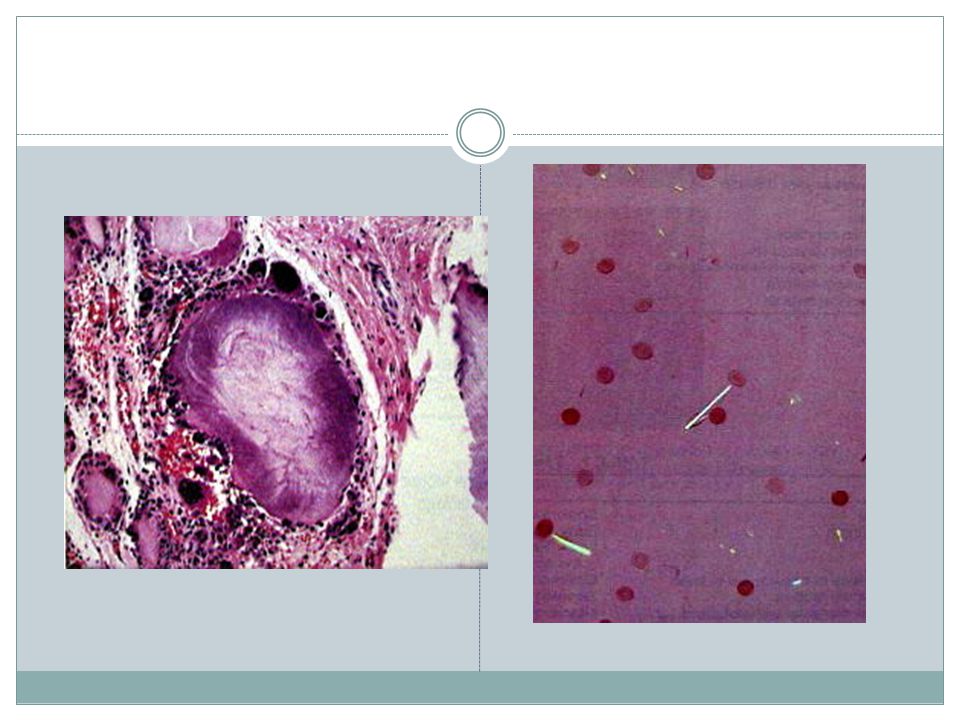
Gout Recurrent episodes of acute arthritis, sometimes accompanied by large crystalline aggregates(tophi) & joint deformity Elevated level of uric acid is an essential component

119
Types Primary( 90% of cases ) - Unknown enzyme defect(85-90% of primary gout) mostly due to overproduction - Known enzyme defect (partial ↓HGPRT) Secondary( 10% of cases ) - ↑nucleic acid turnover(leukemias) - CRF
 Secondary( 10% of cases ) - ↑nucleic acid turnover(leukemias) - CRF.")
120
Clinical features Evolution of gout 1-Asymptomatic hyperuricemia
2-Acute gouty arthritis 3-Intercritical gout 4-Chronic tophaceous gout(arthritis & soft tissue tophi) Gouty nephropathy, renal tubule obstruction , renal stones , tophi
 Gouty nephropathy, renal tubule obstruction , renal stones , tophi.")
121
Major manifestations Acute arthritis
Chronic tophaceous arthritis(deposition on articular cartilage & joint capsule) Persistant chronic inflammation eventually fibrosis of the synovium & erosion of articular cartilage ± fusion of the joint Gouty nephropathy : obstruction of renal tubules by UA crystals , UA renal stones , tophi in the interstitium , scarred & shrunken kidney & CRF
 Persistant chronic inflammation eventually. fibrosis of the synovium & erosion of articular cartilage ± fusion of the joint. Gouty nephropathy : obstruction of renal tubules by UA crystals , UA renal stones , tophi in the interstitium , scarred & shrunken kidney & CRF.")
123
Purine metabolism Complete lack of HGPRT: Lesch-Nyhan syndrome:
Synthesis of purine from nonpurine precursors : Denovo pathway Synthesis of purine nucleotides from free purine bases : Salvage pathway which are catalyzed by two transferases HGPRT & APRT Complete lack of HGPRT: Lesch-Nyhan syndrome: ↑↑↑excretion of UA , severe neurologic dis &MR

128
Acute suppurative arthritis
The most common cause is bacteria Common pathogens:gonococci,staphylococci, streptococci, hemophilus.inf, gram neg rods. Complement deficiency (C5,C6,C7): susceptible to gonococcal arthritis In sickle cell disease : salmonella is important
: susceptible to gonococcal arthritis. In sickle cell disease : salmonella is important.")
129
Lyme disease Involves multiple organ systems
Typically affects large joints such as the knee , shoulder & elbow Early lyme arthritis : synovium resembles early RA & oninoin-skin-like lesions Late dis: extensive erosion of the cartilage in large joints

130
Diseases of skeletal muscle
Basic pathology

135
Skeletal muscle diseases
Neurogenic atrophy Neuromuscular junction disorders(myasthenia gravis) Primary diseases
 Primary diseases.")
136
Primary diseases Myopathy Muscular Dystrophy Congenital
-Ion channel myopathies -Inborn errors of metabolism -Mitochondrial myopathy Toxic Intrinsic exposure( thyroxine ) Extrinsic exposure( alcohol, drugs) Muscular Dystrophy X- linked Autosomal Myotonic dystrophy
 Extrinsic exposure( alcohol, drugs) Muscular Dystrophy. X- linked. Autosomal. Myotonic dystrophy.")
137
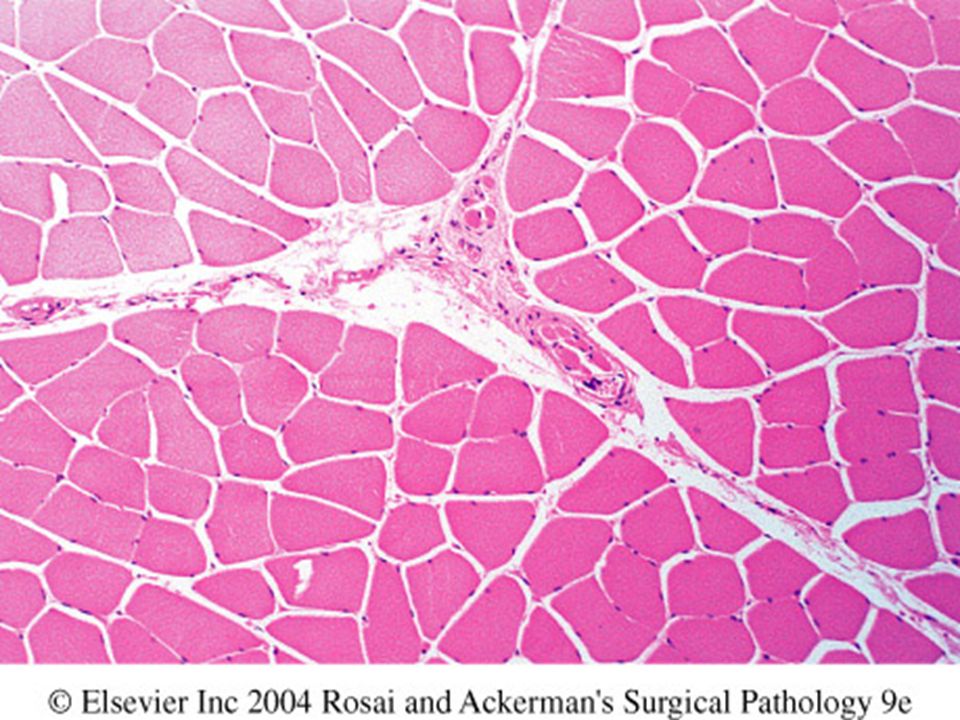
Muscle atrophy Neurogenic atrophy Type 2 myofiber atrophy
The two most common causes: Neurogenic atrophy Type 2 myofiber atrophy disuse atrophy,glucocorticoids, endogenous hypercortisolism

138
Neurogenic atrophy Random atrophy of both fiber types
Angular atrophied fibers Small & later large groups of atrophied fibers Loss of checkerboard pattern with reinnervation (fiber type grouping)
")
139
Reinnervation

140
Werding-Hoffman disease
Markedly atrophic fibers with a rounded cotour Large groups of atrophic fibers Often scattered hyper -trophic fibers

141
Type 2 myofiber atrophy Very nonspecific
Relatively common finding in a muscle BX Most common causes:prolonged steroid therapy,disuse related to prolonged bed rest or joint diseases

142
Type 2 myofiber atrophy angular & atrophic fibers similar to neurogenic atrophy Absence of group atrophy ATPase is essential for DX

143
Myasthenia gravis Acquired autoimmune disorder of neuromuscular transmission Any age Peak age in female 2-3rd decade ,male later F>M Caused by anti-AchR which result in reduced number of AchR by two mechanisms: *internalization & down-regulation of the receptor * blockage of receptors

144
Clinical features Myasthenia gravis Weakness & fatigability of muscles
Cranial muscles,specially lids &extraocular muscles are early involvements(diplopia & ptosis) Weakness increases during repeated use
 Weakness increases during repeated use.")
145
X-linked muscular dystrophy
Defective gene product: dystrophin Clinical features: progressive muscle weakness of proximal limb muscles(early) specially in lower extremity Generalized weakness as the disease progresses Other involvements:cardiomyopathy,mental impairment Cause of death:mainly respiratory failure
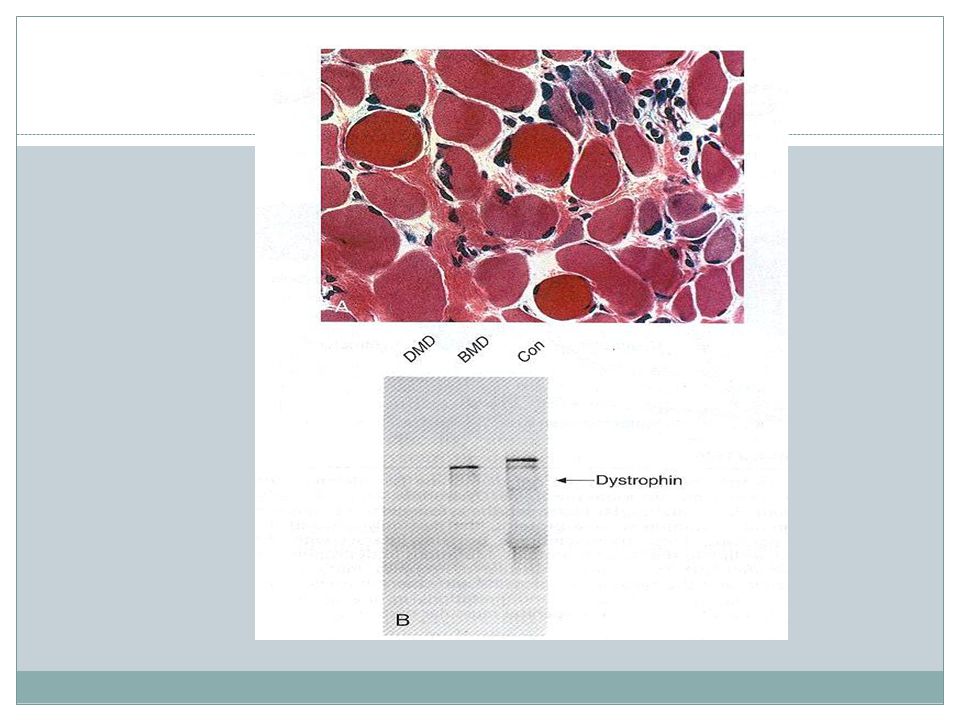
 specially in lower extremity. Generalized weakness as the disease progresses. Other involvements:cardiomyopathy,mental impairment. Cause of death:mainly respiratory failure.")
147
Duchenne muscular dystrophy < 5Y/O
Muscular dystrophies Dystrophin Onset age Clinical course Duchenne muscular dystrophy Absent < 5Y/O Wheelchair dependent by 12 y/o Death at 20 Becker muscular dystrophy Abnormal 5-15 Y/O Walk beyond 15 y/o Most survive into 4thdecade

148
Duchenne’s muscular dystrophy
Marked variation in muscle fiber size Fiber necrosis Myophagia Fiber regeneration Endomysial fibrosis Scattered large hyalinize hypereosinophilic fibers (hypercontracted) Late stage:fiber loss & adipose tissue infiltration
 Late stage:fiber loss & adipose tissue infiltration.")
149
Duchenne’s dystrophy carrier

151
Becker’s muscular dystrophy
Morphology Similar to DMD But fiber necrosis & regenerative changes much less conspicuous than Duchenne’s

155
THE END

Similar presentations





















>")



 normal cell of origin Most are classified.>")














