Download presentation
Presentation is loading. Please wait.

1
Hereditary GI Cancer-a Primer for Medical Oncologists
Ophira Ginsburg, MD, MSc, FRCPC Clinical Lead, Cancer Prevention & Screening Director Familial Oncology Central East Regional Cancer Program March, 2009

2
Objectives HNPCC review
Diagnosis, genetic testing, cancer risks, risk reduction strategies Other Hereditary GI syndromes: FAP, AFAP/MAP, HDGC Criteria for referral to Familial Oncology Programs

3
Hereditary GI Cancer Examples [gene] HNPCC [MLH1, MSH2, MSH6, PMS2]
FAP [APC] AFAP [APC, MUTYH1] FHDG [CDH1]
![Hereditary GI Cancer Examples [gene] HNPCC [MLH1, MSH2, MSH6, PMS2]](http://slideplayer.com/slide/2409318/8/images/3/Hereditary+GI+Cancer+Examples+%5Bgene%5D+HNPCC+%5BMLH1%2C+MSH2%2C+MSH6%2C+PMS2%5D.jpg "FAP [APC] AFAP [APC, MUTYH1] FHDG [CDH1]")
4
Why do genetic testing? To confirm there is a genetic predisposition to cancer To predict which family members are at increased risk of cancer To prevent cancer or detect it early

5
GENETICS SERVICE

6
Genetic Counselling Process
Standard Timeframe 6-8 months Seen <2wks if urgent Courtesy of Lori Van Manen, 2008

7
What Happens During a Genetic Counselling Session?
Risk Assessment Review of hereditary cancer Discussions regarding testing: Pros Obtain information about personal risk Provide incentives for surveillance Clarifies uncertainty Patient empowerment Assists ongoing research Cons Inconclusive results (often) Feelings of guilt or anxiety Family tensions Ethical issues Insurance issues Decreased compliance with screening
 Feelings of guilt or anxiety. Family tensions. Ethical issues. Insurance issues. Decreased compliance with screening.")
8
History of Familial Oncology Programs
1995: counselling and genetic testing became available through research in 1995. 2001: Ministry of Health established recommended referral and testing criteria, and began funding BRCA1/2 testing. Since 2001: HNPCC counselling/testing funded BUT. Local estimates of uptake/referrals given 5-10% CRC caused by HNPCC, ~ 20-25% CRC incident cases *should* be referred. Est: 10% capture so far

9
CRC

10
Lynch Syndrome in Family “G”
Dr. Aldred Scott Warthin, MD, PhD described “Family G” in a 1913 publication based on records ascertained from the University of Michigan hospitals between 1895 and 1913. “Of the 48 descendants of the cancerous grandfather, 17 have died or been operated on for cancer. The preponderance of carcinoma of the uterus (ten cases) and of the stomach (seven cases) is very striking in the family history” Dr. Warthin, 1913. One of the longest family cancer histories ever recorded of a Lynch-syndrome family was reported nearly 100 years ago by Dr. Aldred Scott Warthin. He reviewed the medical histories of cancer patients treated at the University of Michigan hospitals between 1895 and He published his findings on “Family G” - a family characterized at the time by a distinct susceptibility to cancer of the uterus and stomach. Family ‘G’ started with a man born in Plattenhardt, Germany in 1796 and who subsequently immigrated to the US in He had 9 children. As of 2005, 929 known descendants of the original progenitor have been reported Dr. Warthin first published his findings in 1913 – this excerpt was taken from the publication: “Of the 48 descendants of the cancerous grandfather, 17 have died or been operated on for cancer. The preponderance of carcinoma of the uterus (ten cases) and of the stomach (seven cases) is very striking in the family history” Douglas et al. (2005) History of Molecular Genetics of Lynch Syndrome in Family G; JAMA; Vol. 294 (17),
 and of the stomach (seven cases) is very striking in the family history Dr. Warthin, One of the longest family cancer histories ever recorded of a Lynch-syndrome family was reported nearly 100 years ago by Dr. Aldred Scott Warthin. He reviewed the medical histories of cancer patients treated at the University of Michigan hospitals between 1895 and He published his findings on Family G - a family characterized at the time by a distinct susceptibility to cancer of the uterus and stomach. Family ‘G’ started with a man born in Plattenhardt, Germany in 1796 and who subsequently immigrated to the US in He had 9 children. As of 2005, 929 known descendants of the original progenitor have been reported. Dr. Warthin first published his findings in 1913 – this excerpt was taken from the publication: Of the 48 descendants of the cancerous grandfather, 17 have died or been operated on for cancer. The preponderance of carcinoma of the uterus (ten cases) and of the stomach (seven cases) is very striking in the family history Douglas et al. (2005) History of Molecular Genetics of Lynch Syndrome in Family G; JAMA; Vol. 294 (17),")
11
Why HNPCC is important HNPCC/Lynch syndrome- 3 to 5 times more common than FAP Harder to diagnose: many non CRC tumours, families with more “other tumours” than colorectal potential for 1 or 2 prevention of other HNCC cancers: endometrial, urothelial, ovarian?

12
HNPCC -CRC Unique Features
Colorectal cancer: 70% right sided distribution Synchronous, metachronous primaries Pathology: mucinous, poorly differentiated, peri-tumoural lymphocytic infiltration Prognosis: ? Response to chemo?

13
General Population Risk
Cancer General Population Risk HNPCC Risks Mean Age of Onset Colon 5.5% 80% 44 years Endometrium 2.7% 20-60% 46 years Stomach < 1% 11-19% 56 years Ovary 1.6% 9-12% 42.5 years Hepatobiliary tract 2-7% Not reported Urinary tract 4-5% ~ 55 years Small bowel 1-4% 49 years Brain/central nervous system 1-3% ~ 50 years

14
Cancer risk (%) by age 70 CRC 71 39 96 Endometrium 42 61 Ovary 3.4
Type of Cancer MLH1 MSH2 MSH6 CRC Female 71 39 Lower than MLH1/MSH2 Male 96 Endometrium 42 61 Higher than MLH1/MSH2 Ovary 3.4 10.4 specific risk unknown Stomach 2.1 4.3 Small bowel 7.2 4.5 Urinary Tract 1.3 12 Other Extracolonic- 11 Extracolonic -48

15
Family History in HNPCC-more than meets the eye
Lynch and de la Chapelle

18
HNPCC-criteria for testing- beyond Amsterdam*
Revised Amsterdam (ICG-HNPCC, 1999) 3 relatives with CRC or assoc ca (uterine, small bowel, ureter, renal pelvis) 1st degree of the other 2 2 successive generations 1 before age 50 Histological verification Revised Bethesda (Umar et al, 2004) CRC in pt under 50 Synch/metachronous, or other associated ca CRC with MSI under 60 CRC with one or more 1st degree relative (under 50) CRC with 2 or more 1st/ 2nd degree relative (any age) 50% by mutation analysis 15% by mutation analysis *R/O FAP
 3 relatives with CRC or assoc ca (uterine, small bowel, ureter, renal pelvis) 1st degree of the other 2. 2 successive generations. 1 before age 50. Histological verification. Revised Bethesda (Umar et al, 2004) CRC in pt under 50. Synch/metachronous, or other associated ca. CRC with MSI under 60. CRC with one or more 1st degree relative (under 50) CRC with 2 or more 1st/ 2nd degree relative (any age) 50% by mutation analysis. 15% by mutation analysis. *R/O FAP.")
19
2 main classes of CRC: different models of tumourigenesis
Chromosomal instability: 85% distal; aneuploid; APC, p53 K-Ras mutations; more aggressive; prototype FAP “APC= the gatekeeper of CRC” Microsatellite instability: 15% proximal: diploid; MSI, MMR mutations; less aggressive?, prototype HNPCC “MMR genes = caretakers of CRC”

21
Genetic Testing in HNPCC
MSI- microsatellite instability (tumour) IHC- immunohistochemistry (tumour) Germ-line DNA for mutations in one of 3 genes (MLH1, MSH2, MSH6) PMS2 Amsterdam or research lab
 IHC- immunohistochemistry (tumour) Germ-line DNA for mutations in one of 3 genes (MLH1, MSH2, MSH6) PMS2 Amsterdam or research lab.")
23
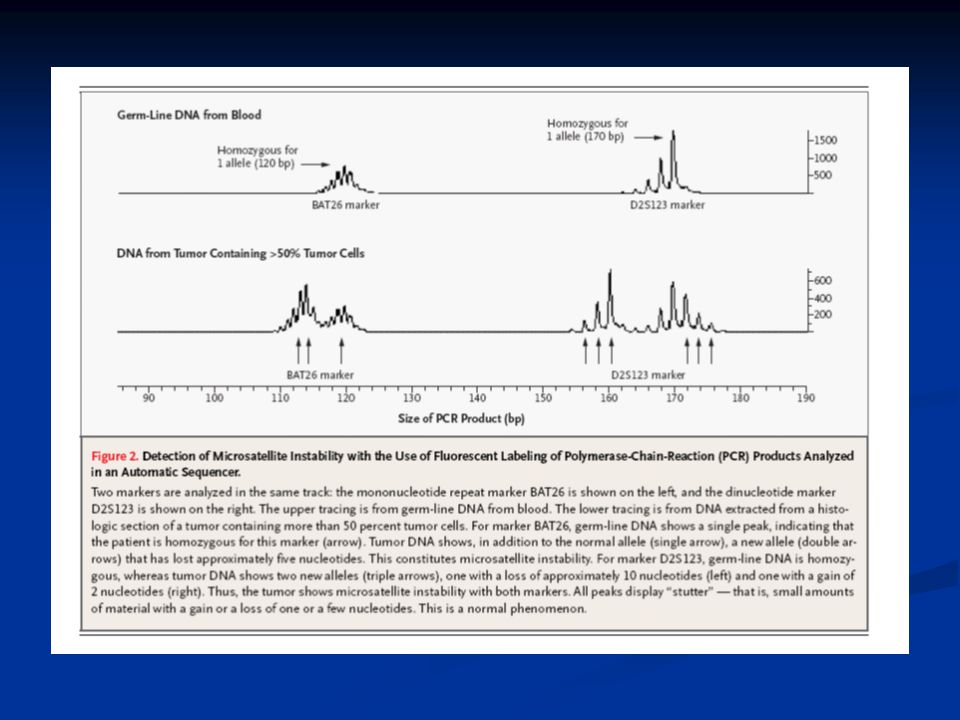
Microsatellite instability
hallmark of tumours in HNPCC Microsatellites: genomic regions with repetitive short DNA sequences (often single nucleotides) prone to mutation during DNA replication Results in elongation or contraction = instability
 prone to mutation during DNA replication. Results in elongation or contraction = instability.")
24
Microsatellite Instability
When DNA polymerase inserts the wrong bases in newly synthesized DNA, the “mismatch repair” enzymes repair the mistake Defects in mismatch repair genes (MLH1, MSH2, MLH6) lead to the mutator phenotype: high frequency microsatellite instability or “MSI-H”
 lead to the mutator phenotype: high frequency microsatellite instability or MSI-H")
26
Gryfe et al, NEJM 2000

27
NEJM May 5,2005 Hampel et al

28
HNPCC: Risk Reduction Options Colorectal
Unaffected: colonoscopy to cecum q 1-2 years Affected w CRC: consider subtotal colectomy at 1st diagnosis d/t risk of 2nd primaries Seminars in Oncology, Oct 2007

29
HNPCC: Risk Reduction Gynecological Cancers
Screening for ovarian ca: CA125 + TVUS jury still out- Lancet Oncology online Mar 09 Screening for endometrial ca: annual TVUS + random endometrial bx (age 30+) few studies: review Lindor et al, JAMA 2006 See also current NCCN guidelines
 few studies: review Lindor et al, JAMA See also current NCCN guidelines.")
30
Surgical Options for Gyne Ca
Prophylactic BSO Ovarian/FT risk reduction > 80-90% nb/ primary peritoneal carcinomatosis ~5-7%) Prophylactic TAH/BSO -less studied but likely >90% RR for endometrial ca in HNPCC mutation carriers Recent meta-analysis for HBOC (BRCA carriers) : J Natl Cancer Inst. 2009;101(2):80-87
 Prophylactic TAH/BSO. -less studied but likely >90% RR for endometrial ca in HNPCC mutation carriers. Recent meta-analysis for HBOC (BRCA carriers) : J Natl Cancer Inst. 2009;101(2):80-87")
32
HNPCC Colorectal Ca Prognosis
many studies: stage-for-stage survival advantage in HNPCC-CRC landmark paper: Gryfe (NEJM 2000): 607 consecutive cases CRC <age 50 17% had MSI-H Multivariate analysis showed a sigificant survival advantage for MSI-H patients versus MSS, independent of ALL other prognostic factors.
: 607 consecutive cases CRC <age % had MSI-H. Multivariate analysis showed a sigificant survival advantage for MSI-H patients versus MSS, independent of ALL other prognostic factors.")
33
The “atypical family history”
Families w multiple cases of non-CRC HNPCC- associated cancers *GU-TCC renal pelvis, bladder, ovarian, squamous cell endometrial, no colorectal cancer in “line of fire” + mutation MSH2 Are the carriers at risk of colorectal ca? YES, and should be screened appropriately

35
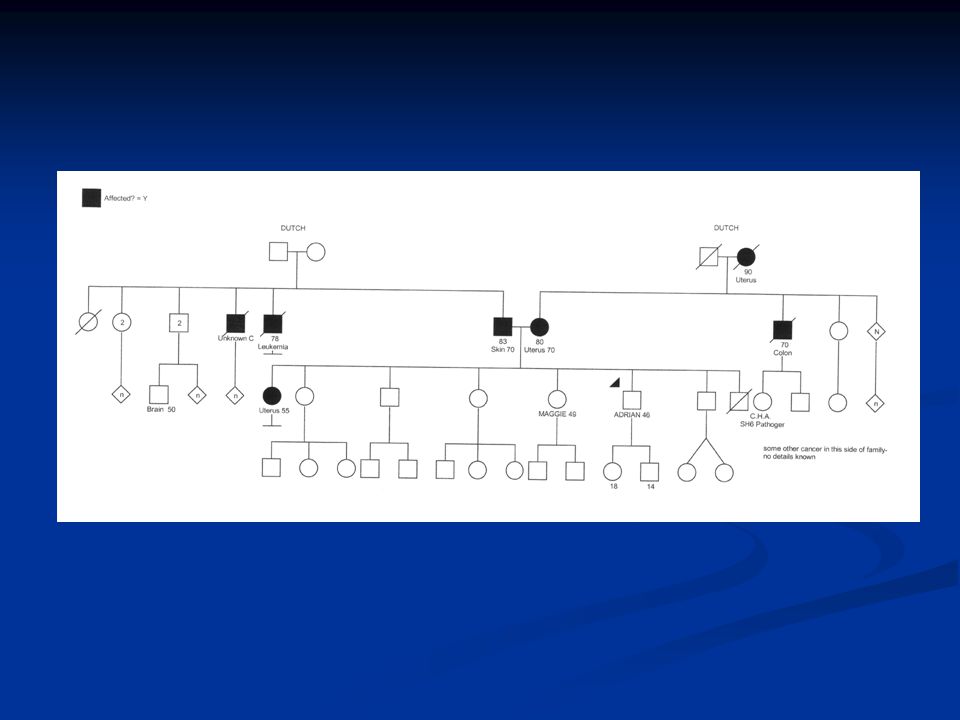
This is one of our Muir-torre families: The mother was referred in 1998 but it was the son who was interested in knowing whether or not the cancers were genetic. Although the family was worked up, no testing was done, although blood had been banked in 1999 and tumour tissue testing was requested in The son was referred directly in 2001 with a newly diagnosed squamous cell carcinoma of the right chest wall. Mother had a transitional cell carcinoma of the renal pelvis at age 60, a Stage 1 ovarian cancer at age 67, and a recurrence of her transitional cell ca in the ureter last year. Proband’s sister had an endometrioid endometrial carcinoma at age 52, treated by TAH and BSO, and this person is currently well. Maternal uncle had pancreatic ca at age 66 and maternal grandmother had ovarian cancer at age 46 and died at age 49. This individual’s sister had bladder cancer, the details of which are unknown. Incidentally, the only case of colon cancer in a relative of the proband is in his father, who had the disease at age 56. He does not have a strong family history to suggest any cancer syndrome, however. Our proband’s sister had MSI/IHC testing and on subsequent germline analysis was found to carry an MSH2 mutation. Mother is a confirmed to be a carrier, as is the other sister. This family took 8 years to work up from the time of referral

38
Some families with Lynch Syndrome present with later onset CRC and may be missed by our current guidelines for genetic testing – guidelines are subject to interpretation and offer we will consider doing MSI/IHC on borderline-eligible families. This proband was referred to us in 1999 and received counseling. At the time testing was offered to the family through paternal aunt (I believe MSI/IHC testing was done on her father’s tumour first). Subsequently, an MSH2 mutation was found. In the meantime, some maternal relatives were part of the OFCCR in Toronto and appeared that the maternal family history was also concerning for HNPCC. Therefore, in 2005 at the request of our proband, germline testing was initiated for the known paternal MSH2 mutation in herself, and MSI/IHC testing was initiated on her maternal cousin(s) with CRC (next slide).
. Subsequently, an MSH2 mutation was found. In the meantime, some maternal relatives were part of the OFCCR in Toronto and appeared that the maternal family history was also concerning for HNPCC. Therefore, in 2005 at the request of our proband, germline testing was initiated for the known paternal MSH2 mutation in herself, and MSI/IHC testing was initiated on her maternal cousin(s) with CRC (next slide).")
39
The first tumour accessed was on the 26 year old dx with rectal cancer at the age of 26. Unfortunately no tumour tissue was available. So, we accessed the tumour tissue of the brother and it showed a deficiency on MLH1. Therefore, we did germline sequencing of the MLH1 gene on our proband in addition to MSH2 carrier testing and voila – and MLH1 mutation was confirmed. Therefore, both of her parents had Lynch Syndrome – different genes involved. This means that our proband’s brother would have a 75% chance of inheriting one, or both of the mutations. Management is a big issue – particularly where the onset of cancers are early. We do not usually test children or adolescents for hereditary adult-onset cancer syndromes, however, the youngest age reported for colon cancer in such a family is 13. In this family, it is 26, so we would start screening with colonoscopy at age 16 and suggest that this be repeated every 1-2 years. This family took about 10 years to work up, and a few genetic counselling centres were involved.

41
FAP- prototype for hereditary cancer
100s to 1000s of adenomatous polyps throughout colon & rectum 100% penetrance without surgery Very early age of onset (polyposis by 20’s most ca by 40s) APC gene chromosome 5
 APC gene chromosome 5.")
42
Risk-reducing surgery in FAP
Sigmoidoscopy: q1-2 ~ age 10-12, genetic testing Colonoscopy: once polyps + annually if colectomy is delayed more than one year Prophylactic Colectomy: recommended- TPC, IRA, IPAA (ileal pouch) Follow-up screening is necessary JCO Oct 1, 2006 ASCO review:
 Follow-up screening is necessary. JCO Oct 1, 2006 ASCO review:")
43
FAP- Desmoids 12-17% of FAP patients
Intra-abdominal 80%, small bowel mesentery >50% (present w SBO) Genotype: APC mutation b/w codons Tend to occur AFTER surgery, high RR, high morbidity Rx: sx for small, well defined desmoids; Tamoxifen, chemo: vinblastine, MTX (RR 40-50%) or for rapidly progressive desmoids per sarcoma protocol (adriamycin, dacarbazine)
 Genotype: APC mutation b/w codons Tend to occur AFTER surgery, high RR, high morbidity. Rx: sx for small, well defined desmoids; Tamoxifen, chemo: vinblastine, MTX (RR 40-50%) or for rapidly progressive desmoids per sarcoma protocol (adriamycin, dacarbazine)")
44
FAP-other tumours Upper GI tumours
80-90% FAP mutation carriers have duodenal or periampullary polyps, of which 36% will develop advanced polyposis 3-5% will develop invasive carcinoma Surveillance: side-viewing endoscopy + bx suspicious yrs Polypectomy for high-grade dysplasia, villous changes, ulceration, > 1 cm size

45
chemoprevention? NSAIDS: level 1 evidence
sulindac, celecoxib, rofecoxib shown to reduce # polyps in FAP, but not proven to reduce cancer incidence or mortality ** long term use as an alternative to sx is NOT recommended + adverse effects Calcium HRT

46
FAP: natural history—revised
Due to improved diagnosis, and prevention/screening for CRC, periampullary cancer and desmoids have become the leading causes of death for FAP(APC) mutation carriers
 mutation carriers.")
47
Other Polyposis Syndromes
AFAP-attenuated familial polyposis polyps, proximal location, later age of onset (1307K allele ~ 6% Ashkenazi Jews, 2x CRC risk) MUTYH1 mutations: “MAP” recessive inheritance -7.5 % of pts w classical phenotype but APC - J. Jass. Pathology Res & Practice (2008) 204:
 MUTYH1 mutations: MAP recessive inheritance % of pts w classical phenotype but APC - J. Jass. Pathology Res & Practice (2008) 204:")
48
Attenuated FAP

49
“MAP” MYH-associated polyposis coli
Nielsen et al, Journal of Medical Genetics 2005

50
Hereditary Diffuse Gastric Cancer
E-cadherin CDH1 gene 70% risk of diffuse gastric ca 40% risk of lobular breast ca Prophylactic total gastrectomy ?screening chromoendoscopy Lynch et al, Cancer 2008 Jun 15;112(12):
:")
51
Objectives HNPCC review
Diagnosis, genetic testing, cancer risks, risk reduction strategies Other Hereditary GI syndromes: FAP, AFAP/MAP, HDGC Criteria for referral to Familial Oncology Programs

Similar presentations