Download presentation
Presentation is loading. Please wait.

1
İSCHAEMİC HEART DİSEASE
Prof Dr. Rasim ENAR Istanbul University Cerrahpasa Medical Faculty Department Cardiology

2
Myocardial ischaemia result of acute or persistently (chronic) decrease myocardial blood flow due to narrowed coronary lumen diameter, mainly caused by coronary Atherosclerosis. Mechanisms: 1. While patient has stable coronay obstructıon due to sinificant plaque stenosis ( ≥ %50 lumen diameter narrowing) mostly remain asyptomatic in the resting conditıons. At this situatıon coronary flow never respond (augmented) to increased myocardial oxygen demand (strenous exertıon, emotıonal stress); so myocardial ischaemia occurred, as a result of demand – supply dismatch; clinically termed Chronic Coronary artery disease (CAD) or stable angina pectoris. 2. On the other hand, patient who has previously near normal coronary flow with insignificant obstructive plaque (<%50 luminal narrowing); when coronary flow sudden decrease below the critcal level ( supply not matching resting demand ) from result of abrupt trhrombosıs uperimposed over plaque disruptıon, ultimately acute myocardial ischeamia developed, İn clinical practice called Acute coronary syndromes (ACS). Main clinical presentatıons of ACS are; regardless amount of myocardial injury are sudden cardiac death, symptomatic or asymptomatic acute myocardial ischaemia.
 mostly remain asyptomatic in the resting conditıons. At this situatıon coronary flow never respond (augmented) to increased myocardial oxygen demand (strenous exertıon, emotıonal stress); so myocardial ischaemia occurred, as a result of demand – supply dismatch; clinically termed Chronic Coronary artery disease (CAD) or stable angina pectoris. 2. On the other hand, patient who has previously near normal coronary flow with insignificant obstructive plaque (<%50 luminal narrowing); when coronary flow sudden decrease below the critcal level ( supply not matching resting demand ) from result of abrupt trhrombosıs uperimposed over plaque disruptıon, ultimately acute myocardial ischeamia developed, İn clinical practice called Acute coronary syndromes (ACS). Main clinical presentatıons of ACS are; regardless amount of myocardial injury are sudden cardiac death, symptomatic or asymptomatic acute myocardial ischaemia.")
3
ATHEROSCLEROSIS: Atherosclerosos İs inflamatory disease of the arterial wall that underlies many of the common causes of cardiovascular morbidity and mortality; including myocardial infarction (MI), peripheral vascular disease (and cerebrovascular disease). Coronary artery disease (CAD) causes severe disability and more death than any other disease (including cancer). Atherosclerosıs develops over the course of 50 years, beginning in the early teenage years. Risk Factors for (atherosclerotic ) CAD: Major independent RF: Advanced age; male gender, postmenopausal women. Tobacco smoking, Diabetes mellitus, Elevated total and low-density lipoprotein cholesterol (LDL), Low high density lipoprotein cholesterol (HDL), Hypertensıon. METs (hypertensıon, abdominal obesity, dislipidemia, insülin resistan/diabetes). Predisposing RF: Abdominal obesity; Ethnic characteristics; Family history of premature Coronary heart disease ( first degree realatives; <55 years for men, <65 years for women). Obesity; physical inactivity; psychosocial factors.
, peripheral vascular disease (and cerebrovascular disease). Coronary artery disease (CAD) causes severe disability and more death than any other disease (including cancer). Atherosclerosıs develops over the course of 50 years, beginning in the early teenage years. Risk Factors for (atherosclerotic ) CAD: Major independent RF: Advanced age; male gender, postmenopausal women. Tobacco smoking, Diabetes mellitus, Elevated total and low-density lipoprotein cholesterol (LDL), Low high density lipoprotein cholesterol (HDL), Hypertensıon. METs (hypertensıon, abdominal obesity, dislipidemia, insülin resistan/diabetes). Predisposing RF: Abdominal obesity; Ethnic characteristics; Family history of premature Coronary heart disease ( first degree realatives; <55 years for men, <65 years for women). Obesity; physical inactivity; psychosocial factors.")
4
Pathophysıology of Atherosclerosıs:
The normal vessel wall and endothelial function: A normal, healthy artery comprises: 1- Endothelium – a monolayer of endothelial cells and its basement membrane cover inner surface of vessel lumen. 2- Media – concentric layers of smooth muscle cells, elastin fibres and extracellular matrix. 3- Surrounding Adventitia of connective tissue. The endothelial cell layer maintaining vascular homeostasis. Endothelial cells provide a functional link between blood in the lumen and the vessel wall. They transduce many physiological stimuli, that have effects on blood cells, endothelium itself and on neighbouring smooth muscle cells. Nitric oxide (NO) is one of the most important signalling molecules produced by the endothelium. NO was originally identified as ‘endothelium-derived relaxing factor’. In normal blood vessels, NO is generated by the enzyme endothelial nitric oxide synthase (eNOS), which is present in endothelial cells.
 is one of the most important signalling molecules produced by the endothelium. NO was originally identified as ‘endothelium-derived relaxing factor’. In normal blood vessels, NO is generated by the enzyme endothelial nitric oxide synthase (eNOS), which is present in endothelial cells.")
5
Endothelial dysfunction in Atherosclerosis:
The overall effects of NO on the vessel wall are to inhibit the processes that contribute to early atherosclerosis . İn diseased artery , early and characteristic feature of is abnormal function of the endothelium (endotelial dysfunctıon); characterized by loss of endothelial NO bioactivity and associated activation of the endothelium; resulting in predisposition to thrombosis and inflammatory cell adhesion and recruitment . * İn the following conditions, endothelial dysfunction can be detected before macroscopic atherosclerosis is visible: • Hypercholesterolaemia • Smoking • Hypertension • Heart failure. * Key point: İn all these patients with atherosclerosis, arteries are tructurally normal. Clinically, endothelial dysfunction manifests as abnormalities in NO-mediated vascular responses: (for example: lack of dilatation, or paradoxical constriction of coronary artery segments in response to intracoronary infusion of acetylcholine).
; characterized by loss of endothelial NO bioactivity and associated activation of the endothelium; resulting in predisposition to thrombosis and inflammatory cell adhesion and recruitment . * İn the following conditions, endothelial dysfunction can be detected before macroscopic atherosclerosis is visible: • Hypercholesterolaemia. • Smoking. • Hypertension. • Heart failure. * Key point: İn all these patients with atherosclerosis, arteries are tructurally normal. Clinically, endothelial dysfunction manifests as abnormalities in NO-mediated vascular responses: (for example: lack of dilatation, or paradoxical constriction of coronary artery segments in response to intracoronary infusion of acetylcholine).")
6
Progressive molecular pathology and Clinical event:
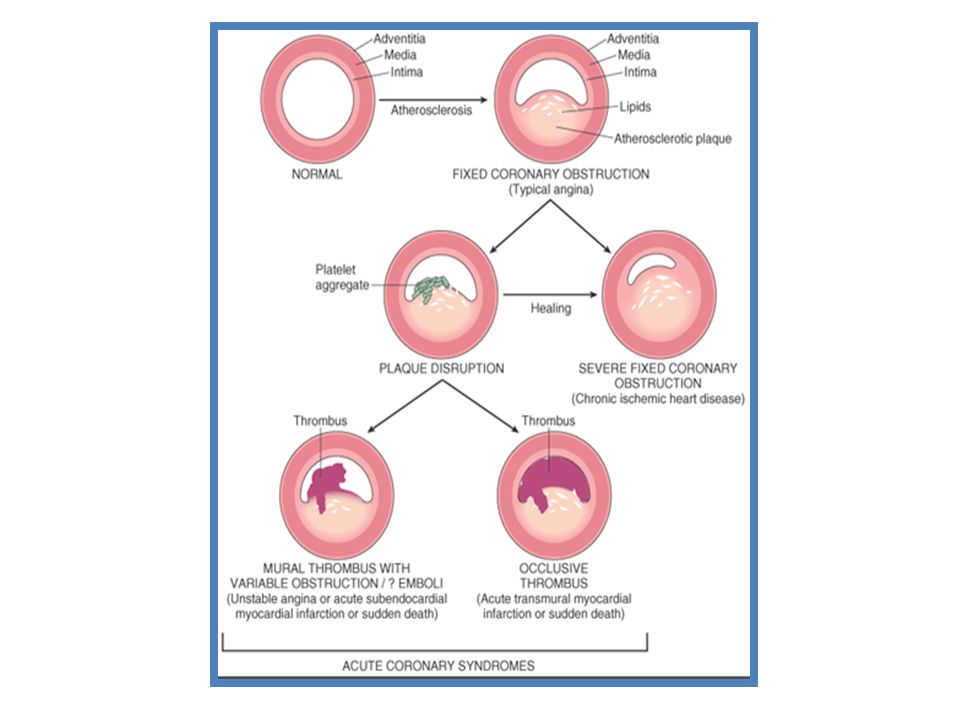
ATHEROSCLEROSİS. The causes of this process appear to be Lipid retention, oxidation, and modification, which provoke chronic inflammation at susceptible sites in the walls of all major conduit arteries. Initial visible lesion Fatty streaks evolve into fibrous plaques, some of develop into forms vulnerable prone to rupture, causing thrombosis or stenosis. CAD is almost always due to atheromatous narrowing and subsequent occlusion of the vessel. Early atheroma is present from young and progress lifelong. A mature plaque is composed of two constituents, each associated with a particular cell population: 1) The lipid core is (“lipid gruels”) formed mainly released from necrotic“foam cells”( monocyte derived macrophages), which migrate into the intima and ingest lipids. 2) The connective tissue matrix is derived from smooth muscle cells, which migrate from the media into the intima, where they proliferate and change their phenotype to form a fibrous capsule around the lipid core. When a plaque produces a > 50% diameter stenosis (or > 75% reduction in cross sectional area), reduced blood flow through the coronary artery during exertion may lead to Angina. Acute coronary events usually arise when acute thrombus formation follows disruption of a plaque. In Acute myocardial infarction, occlusion is more complete than in Unstable angina pectoris (USAP), where arterial occlusion is usually subtotal. Downstream embolism of thrombus may also produce microinfarcts in UAP.
 The lipid core is ( lipid gruels ) formed mainly released from necrotic foam cells ( monocyte derived macrophages), which migrate into the intima and ingest lipids. 2) The connective tissue matrix is derived from smooth muscle cells, which migrate from the media into the intima, where they proliferate and change their phenotype to form a fibrous capsule around the lipid core. When a plaque produces a > 50% diameter stenosis (or > 75% reduction in cross sectional area), reduced blood flow through the coronary artery during exertion may lead to Angina. Acute coronary events usually arise when acute thrombus formation follows disruption of a plaque. In Acute myocardial infarction, occlusion is more complete than in Unstable angina pectoris (USAP), where arterial occlusion is usually subtotal. Downstream embolism of thrombus may also produce microinfarcts in UAP.")
7
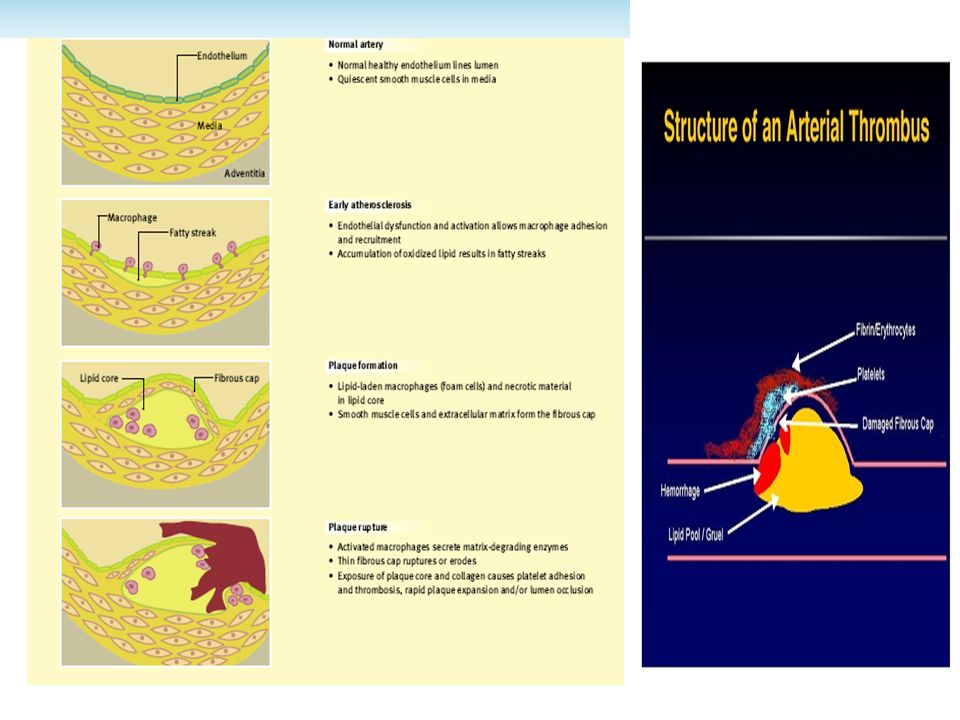
Progression of atheromatous plaque from initial lesion to complex and ruptured plaque ■ (Left): Schematic representation of normal coronary artery wall (top) and development of atheroma (bottom).
: Schematic representation of normal coronary artery wall (top) and development of atheroma (bottom).")
9
ATHEROTHOMBOSIS and High Risk Plaque:
Atherothrombosis is a complex disease in which cholesterol deposition, inflammation, and thrombus formation play a major role. The atherosclerotic disease is asymptomatic during a long period and dramatically changes its course when complicated by thrombosis. Rupture of high-risk vulnerable plaques is responsible for coronary thrombosis; which it is the the main cause of unstable angina, acute myocardial infarction, and sudden cardiac death. In addition to rupture, plaque erosion may also lead to occlusive thrombosis and acute coronary events. Exposure of thrombogenic substrate as resultant of plaque rupture (Virchow triad) is a key factor in determining thrombogenicity at the local arterial site.
 is a key factor in determining thrombogenicity at the local arterial site.")
10
THE VİRCHOW TRİAD OF THROMBOGENİCİTY:
1) Local vessel wall substrates Atherosclerosis: Degree of plaque disruption (i.e., erosion, ulceration) Vessel wall inflammation: Components of plaque (i.e., lipid core) Macrophages and generation of microparticles (i.e., tissue factor content) Post-interventional vessel wall injury: Plaque disruption after percutaneous transluminal coronary angioplasty, atherectomy, or stenting Injury of smooth-muscle cells (i.e., rich in thrombin) 2) Rheology High shear stress: Severe stenosis (i.e., change in geometry with plaque disruption, residual thrombus) Vasoconstriction (i.e., serotonine, thromboxane A2, thrombin, dyfunctional endothelium) Oscillatory shear stress: Bifurcation of arteries, plaque irregularities Post-intervention slow blood flow/local stasis (i.e., dissecting aneurysm). 3) Systemic factors of the circulating blood Metabolic or hormonal factors Dyslipoproteinemia [triglycerides, increased low-density lipoprotein or oxidized low-density lipoprotein cholesterol, decreased high density lipoprotein cholesterol, lipoprotein(a)] Diabetes mellitus (i.e., glycosylation) Catecholamines (i.e., smoking, stress, cocaine use) Renin-angiotensin system (i.e., high-renin hypertension) Plasma variables of hemostasis: Tissue factor, factor VII, factor VII, fibrinogen, thrombin generation (fragments 1 and 2), thrombin activity (fibrinopeptide A), plasminogen activator inhibitor-1, tissue plasminogen activator. Infectious (i.e., Chlamydia pneumoniae, cytomegalovirus, Helicobacter pylori) and cellular blood elements (i.e., monocytes and white blood cells)
 Local vessel wall substrates. Atherosclerosis: Degree of plaque disruption (i.e., erosion, ulceration) Vessel wall inflammation: Components of plaque (i.e., lipid core) Macrophages and generation of microparticles (i.e., tissue factor content) Post-interventional vessel wall injury: Plaque disruption after percutaneous transluminal coronary angioplasty, atherectomy, or stenting. Injury of smooth-muscle cells (i.e., rich in thrombin) 2) Rheology. High shear stress: Severe stenosis (i.e., change in geometry with plaque disruption, residual thrombus) Vasoconstriction (i.e., serotonine, thromboxane A2, thrombin, dyfunctional endothelium) Oscillatory shear stress: Bifurcation of arteries, plaque irregularities. Post-intervention slow blood flow/local stasis (i.e., dissecting aneurysm). 3) Systemic factors of the circulating blood. Metabolic or hormonal factors. Dyslipoproteinemia [triglycerides, increased low-density lipoprotein or oxidized low-density lipoprotein cholesterol, decreased high density. lipoprotein cholesterol, lipoprotein(a)] Diabetes mellitus (i.e., glycosylation) Catecholamines (i.e., smoking, stress, cocaine use) Renin-angiotensin system (i.e., high-renin hypertension) Plasma variables of hemostasis: Tissue factor, factor VII, factor VII, fibrinogen, thrombin generation (fragments 1 and 2), thrombin activity (fibrinopeptide A), plasminogen activator inhibitor-1, tissue plasminogen activator. Infectious (i.e., Chlamydia pneumoniae, cytomegalovirus, Helicobacter pylori) and cellular blood elements (i.e., monocytes and white blood cells)")
11
Cross-sectioned coronary artery containing a ruptured plaque with a non-occlusive platelet-rich thrombus superimposed: The actual defect in the fibrous cap is not seen in this section but is located nearby, documented by the presence of extravasated radiographic contrast medium (postmortem coronary angiography) in the soft, lipid-rich core just beneath the thin, inflamed fibrous cap. ■ Trichrome stain, rendering thrombus red, collagen blue, and lipid colorless. * Three major determinants of plaque’s vulnerability to rupture: (1) The size and consistency of the atheromatous core; (2) Thickness the fibrous cap covering the core; (3) İnflamatıon and repair within the cap.
 The size and consistency of the atheromatous core; (2) Thickness the fibrous cap covering the core; (3) İnflamatıon and repair within the cap..")
12
Plaque vulnerability, disruption, and thrombosis: Anatomical changes leading to acute coronary syndrome and subsequent plaque remodeling. An element of vasoconstriction is usually present.

14
ACUTE CORONARY SYNDROMES:

15
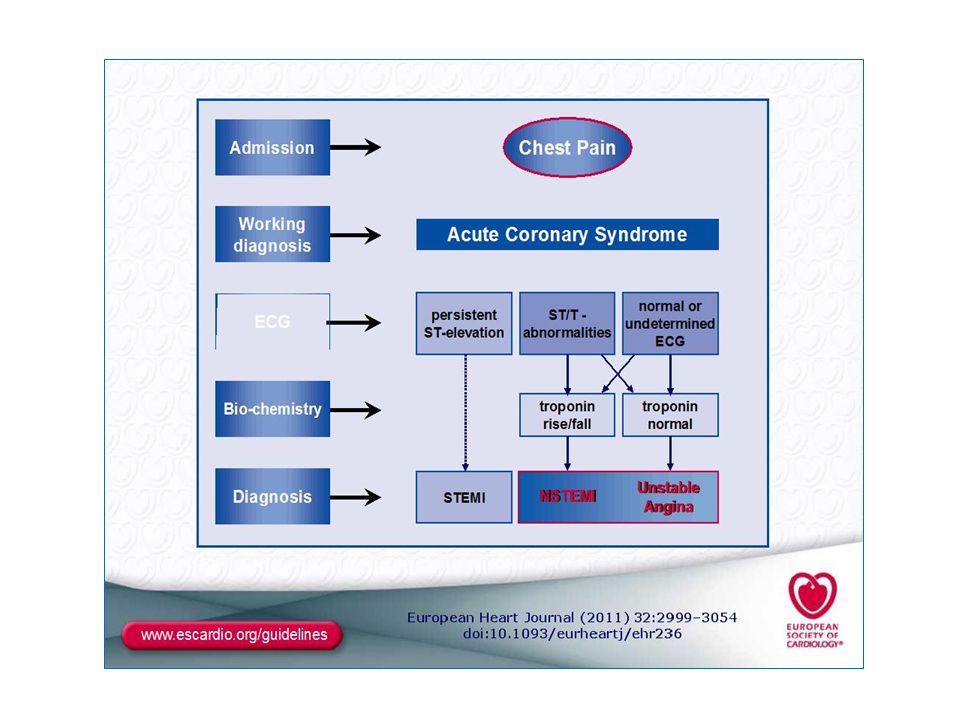
Definitıon: ACS, termed; symptoms resulting from Acute myocardial ischaemia due to acute atherothrombotic events. Presentatıon of ACS includes wide range of clinical condition; from asymptomatic myocardial infarctıon to sudden cardiac death, and also typically unstable angina, ST segment elevatıon , non-ST segment elevatıon Mİ. The current nomeclature divide ACS into 2 main subgroups by ECG findings (ST- segment, T- wave). This clasificatıon also guided the primary therapy.
. This clasificatıon also guided the primary therapy.")
16
The classification of ACS (based on the electrocardiogram):
Patients with prolonged acute chest pain and persistent (>20 min) ST-segment elevation is termed ST-elevation ACS (STE-ACS) and generally reflects an acute total coronary occlusion. Ultimately, most of these patients will develop an ST-elevation MI (STEMI). Because of major luminal pathology are total occlusıon by fibrin-rich thrombus, this group of patients should be undergone reperfusıon therapy, as soon as possible. The therapeutic objective is to achieve rapid, complete, and sustained reperfusion by primary percutaneous coronary interventıon (PCI) or fibrinolytic therapy(FLT). 2. Patients with acute prolonged chest pain with rather than persistent ST-segment elevation; transient ST-segment depression or T-wave inversion, flat T waves, pseudo-normalization of T waves, or no ECG changes at presentation . The initial strategy in these patients is to alleviate ischaemia, symptoms,and stop ongoing Mİ. At this group of patients, because of thrombus non-occlusive and platelet- rich, İntensive antithrombotics (particularly antiplatelets, antithrombins and combinatıon) mainstay of therapy, not treated with fibrinolytic therapy. At presentation, the working diagnosis of non-ST-elevation ACS (NSTE-ACS), based on the measurement of troponins, will be further qualified as non-ST-elevation MI (NSTEMI) or unstable angina (USAP). İn Patients with non-ST-segment elevatıon at presentıon ECG; İf there is biochemical evidence of myocardial injury (elevated btroponin and CK- MB) termed NSTEMİ, and in the absence of myocardial injury biomaker is termed USAP. However, slight cTn elevatıon was found some in USAP patients whose early recurrence of ischaemic events risk was increased ( termed High- risk USAP).
 ST-segment elevation is termed ST-elevation ACS (STE-ACS) and generally reflects an acute total coronary occlusion. Ultimately, most of these patients will develop an ST-elevation MI (STEMI). Because of major luminal pathology are total occlusıon by fibrin-rich thrombus, this group of patients should be undergone reperfusıon therapy, as soon as possible. The therapeutic objective is to achieve rapid, complete, and sustained reperfusion by primary percutaneous coronary interventıon (PCI) or fibrinolytic therapy(FLT). 2. Patients with acute prolonged chest pain with rather than persistent ST-segment elevation; transient ST-segment depression or T-wave inversion, flat T waves, pseudo-normalization of T waves, or no ECG changes at presentation . The initial strategy in these patients is to alleviate ischaemia, symptoms,and stop ongoing Mİ. At this group of patients, because of thrombus non-occlusive and platelet- rich, İntensive antithrombotics (particularly antiplatelets, antithrombins and combinatıon) mainstay of therapy, not treated with fibrinolytic therapy. At presentation, the working diagnosis of non-ST-elevation ACS (NSTE-ACS), based on the measurement of troponins, will be further qualified as non-ST-elevation MI (NSTEMI) or unstable angina (USAP). İn Patients with non-ST-segment elevatıon at presentıon ECG; İf there is biochemical evidence of myocardial injury (elevated btroponin and CK- MB) termed NSTEMİ, and in the absence of myocardial injury biomaker is termed USAP. However, slight cTn elevatıon was found some in USAP patients whose early recurrence of ischaemic events risk was increased ( termed High- risk USAP).")
17
Acute Coronary Syndromes MYOCARDİAL İNFARCTION
Superfıcıal erosıon Ruptured Fibrous cap Total occlusıon Nonocclusıve Critical stenosıs Platelet- rich Thrombus Fibrin- rich Thrombus Non-ST- Elevatıon ST- Elevatıon cTn↑ CK/MB↑ CK-MB- cTn↑ CK-MB - cTn - NSTEMI MYOCARDİAL İNFARCTION NQMI Q MI USAP + High- RİSK

18
Clinical Features of Myocardial Ischaemia and Infarction:
Onset of myocardial ischaemia is the initial step in the development of MI , results from an imbalance between oxygen supply and demand. Myocardial ischaemia in clinical setting can usually be identified from the patient’s history and the ECG. Suggested ischaemic symptoms include various combinations of chest, upper extremity, mandibular pain or epigastric discomfort (with exertion or at rest) b) ischaemia equivalent symptoms are not rare; such as dyspnoea or fatigue, dizziness, lightheadedness. c) The discomfort associated with acute MI usually lasts >20 min. d) Often, the discomfort is diffuse— (not localized, not positional,and not affected by movement of the region)— and it may be accompanied by diaphoresis, nausea or syncope. However, these symptoms are not specific for myocardial ischaemia. Accordingly, they may be misdiagnosed and attributed to gastrointestinal, neurological, pulmonary or musculoskeletal disorders. e) MI may occur with atypical symptoms—such as palpitations or cardiac arrest— or even without symptoms (silent). For example in women, the elderly, diabetics, or post-operative and critically ill patients. Careful evaluation of these patients is advised.
 b) ischaemia equivalent symptoms are not rare; such as dyspnoea or fatigue, dizziness, lightheadedness. c) The discomfort associated with acute MI usually lasts >20 min. d) Often, the discomfort is diffuse— (not localized, not positional,and not affected by movement of the region)— and it may be accompanied by diaphoresis, nausea or syncope. However, these symptoms are not specific for myocardial ischaemia. Accordingly, they may be misdiagnosed and attributed to gastrointestinal, neurological, pulmonary or musculoskeletal disorders. e) MI may occur with atypical symptoms—such as palpitations or cardiac arrest— or even without symptoms (silent). For example in women, the elderly, diabetics, or post-operative and critically ill patients. Careful evaluation of these patients is advised.")
19
CHEST PAİN / DİSCOMFORT İN AMİ PATİENT:
Usually lasts more than 20 minutes and often persists for several hours. The pain of Mİ however can last in 15 minutes; occasionally fatal infarction is developed in by only a few minutes of severe pain or even unheralded cardiac arrest. Quality: Patients with prior angina pectorıs usually describe their discomfort of AMİ is being similar in quality, but more severe. While substernal pressure is unlikely to be of superficial origin, Most of the patient said: ”Elephant sitting on my chest” (or pressure, heaviness) or the clenched fist sign”. Common locatıon: Smilar that of described for angina pectoris. The pain is usually located in retrosternal area, it often radiates to the neck, jaw, left shoulder and inner aspect of left arm. Uncommon locatıon: Are similiar to that described for angina pectoris.The pain may felt only in the lower jaw,left or right shoulder, inner or outer aspect of the left or right upper arm, left or right elbow, left or right wrist. Size of painful area: The size painful area on the chest is the clenched fist or larger. Duratıon of the pain: The pain of AMİ usually last longer than 30 minutes. After resolutıon of the most intense pain, patient describe less intense heaviness or ache that may last for hours. Pain of AMİ; pain onset is with low İntensity and in following minutes gradually increase and reaching peak in a hour and persisted several hours until reperfused. Usually not respond sublingual NTG. Patient filing doom.
 or the clenched fist sign . Common locatıon: Smilar that of described for angina pectoris. The pain is usually located in retrosternal area, it often radiates to the neck, jaw, left shoulder and inner aspect of left arm. Uncommon locatıon: Are similiar to that described for angina pectoris.The pain may felt only in the lower jaw,left or right shoulder, inner or outer aspect of the left or right upper arm, left or right elbow, left or right wrist. Size of painful area: The size painful area on the chest is the clenched fist or larger. Duratıon of the pain: The pain of AMİ usually last longer than 30 minutes. After resolutıon of the most intense pain, patient describe less intense heaviness or ache that may last for hours. Pain of AMİ; pain onset is with low İntensity and in following minutes gradually increase and reaching peak in a hour and persisted several hours until reperfused. Usually not respond sublingual NTG. Patient filing doom.")
20
AMİ Associated Symptoms :
• Diaphoresis, cold clammy skin, and apprehension (however, all of these symptoms may be absent). • Shortness of breath, nausea, vomiting, dizziness. • Women with acute MI often reveal atypical symptoms with low levels of chest pain or absence of pain. Shortness of breath (57.9%), weakness (54.8%), and fatigue (42.9%). • Presyncope and rarely syncope may occur owing to bradyarrhythmias, especially inferior MI. Because of bradyarrithmias due to atrioventricular , sinoatrial conductıon blocks and excessive vagal stimulatıon nausea, presyncope, syncope vomiting, dizziness more characteristic for inferior AMİ. Painless infarcts (in about 10% of patients), especially in diabetics or the elderly. Physical Signs •Patient appears apprehensive (feeling doom), restless, anxious, cold, clammy. Area of chest pain may be indicated with a clenched fist (Levine sign). Chest pain never located and precipitated by two fingers.
. • Shortness of breath, nausea, vomiting, dizziness. • Women with acute MI often reveal atypical symptoms with low levels of chest pain or absence of pain. Shortness of breath (57.9%), weakness (54.8%), and fatigue (42.9%). • Presyncope and rarely syncope may occur owing to bradyarrhythmias, especially inferior MI. Because of bradyarrithmias due to atrioventricular , sinoatrial conductıon blocks and excessive vagal stimulatıon nausea, presyncope, syncope vomiting, dizziness more characteristic for inferior AMİ. Painless infarcts (in about 10% of patients), especially in diabetics or the elderly. Physical Signs. •Patient appears apprehensive (feeling doom), restless, anxious, cold, clammy. Area of chest pain may be indicated with a clenched fist (Levine sign). Chest pain never located and precipitated by two fingers.")
21
Common locations of cardiac pain: ■ Language of hands. ■ “Levine sign”.

22
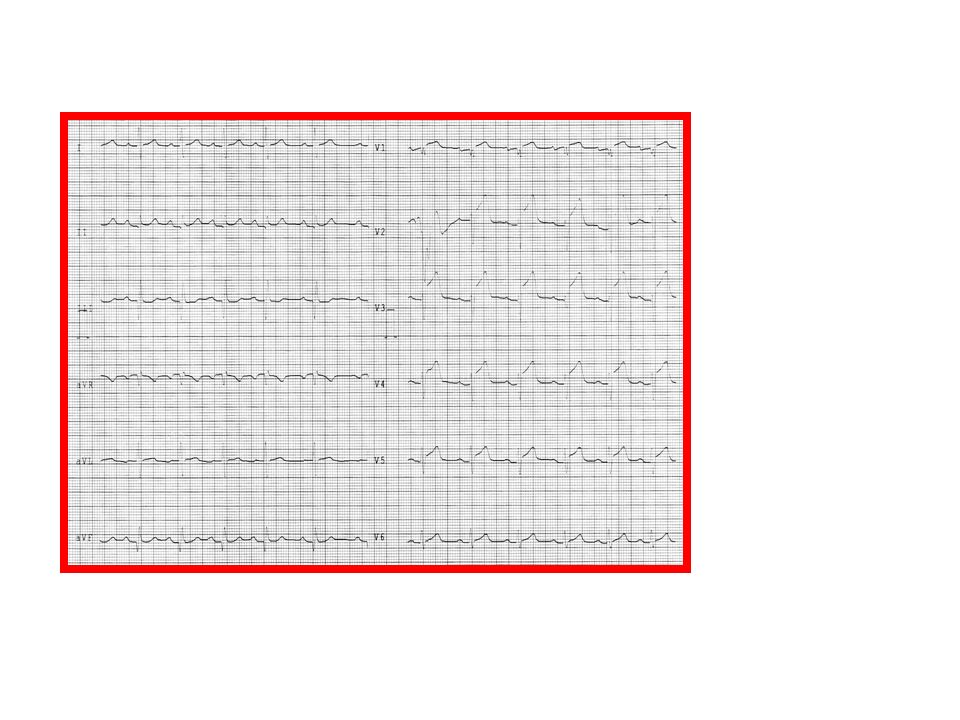
Acute Myocardial ischemia:
ECG Manifestatıon of Acute Myocardial ischemia:

26
Use of Cardiac Markers in ACS
URL = 99th %tile of Reference Control Group 1 2 5 10 20 50 Cardiac troponin after “classical” AMI CK-MB after AMI Cardiac troponin after “microinfarction” Multiples of the URL Upper reference limit Days After Onset of AMI Myocardial injury is detected when blood levels of sensitive and specific biomarkers such as cTn or the MB fraction of creatine kinase (CKMB) are increased. Blood samples for the measurement of cTn should be drawn on first assessment (after >6-8h of AMİ onset).
 are increased. Blood samples for the measurement of cTn should be drawn on first assessment (after >6-8h of AMİ onset).")
27
ACUTE ST- SEGMENT ELEVATION Mİ (STEMİ):
ST-segment elevatıon or new LBBB in the setting of symptoms consistent with acute myocardial ischemia and detected evidence of myocardial necrosis. Practical Formula: (“ECG, + Symptoms, + BioNecrosis = Mİ”). İmportance of Mİ: Worldwide, coronary artery disease (CAD) is the single most frequent cause of death. Over seven million people every year die from CAD, accounting for 12.8% of all deaths. Every sixth man and every seventh woman in Europe will die from myocardial infarction. The mortality of STEMI is influenced by many factors: (age, Killip class, time delay to treatment, mode of treatment, Mİ locatıon history of prior myocardial infarction, diabetes mellitus, renal failure,and number of diseased coronary arteries, LV ejection fraction). The in-hospital mortality varies between 6% and 14%. Decline acute and long-term mortality following STEMI in parallel with greater use of reperfusion therapy, primary PCI, modern antithrombotic therapy and secondary prevention treatments. Still, mortality remains substantial with approximately 12% of patients dead within 6 months,
. İmportance of Mİ: Worldwide, coronary artery disease (CAD) is the single most frequent cause of death. Over seven million people every year die from CAD, accounting for 12.8% of all deaths. Every sixth man and every seventh woman in Europe will die from myocardial infarction. The mortality of STEMI is influenced by many factors: (age, Killip class, time delay to treatment, mode of treatment, Mİ locatıon history of prior myocardial infarction, diabetes mellitus, renal failure,and number of diseased coronary arteries, LV ejection fraction). The in-hospital mortality varies between 6% and 14%. Decline acute and long-term mortality following STEMI in parallel with greater use of reperfusion therapy, primary PCI, modern antithrombotic therapy and secondary prevention treatments. Still, mortality remains substantial with approximately 12% of patients dead within 6 months,")
28
Pathological characteristics of AMİ:
MI is defined in pathology as myocardial cell death due to prolonged ischaemia. After the onset of myocardial ischaemia, histological cell death is not develop, immediately, takes a defifinite period of time to cell-death (as little as 20 min, or less). It takes several hours before myocardial necrosis can be identified by macroscopic or microscopic post-mortem examination. Complete necrosis of myocardial cells at risk requires at least 2–4 h, or longer; depending on the presence of collateral circulation to the ischaemic zone, persistent or intermittent coronary occlusion, the sensitivity of the myocytes to ischaemia, The entire process leading to a healed infarction usually takes at least 5–6 weeks. Definitıon of Myocardial İnfarctıon: The term of Mİ should be used when there is evidence of myocardial necrosis in a clinical setting onsistent with acute myocardial ischemia. Under this conditıon one of the following critera meet the diagnosis of Mİ: Detectıon of a rise an/or fall of cardiac biomarkers (cTn) with at least one value above 99th percantile of URL and with at least one of the following: -+ ST-T changes or new LBB. -+ Development pathological Q wave . -+İmaqging new loss of viable myocardium or regional wall motıon abnormality. -+ İdentificatıon of intracoronary thrombus by angiography.
. It takes several hours before myocardial necrosis can be identified by macroscopic or microscopic post-mortem examination. Complete necrosis of myocardial cells at risk requires at least 2–4 h, or longer; depending on the presence of collateral circulation to the ischaemic zone, persistent or intermittent coronary occlusion, the sensitivity of the myocytes to ischaemia, The entire process leading to a healed infarction usually takes at least 5–6 weeks. Definitıon of Myocardial İnfarctıon: The term of Mİ should be used when there is evidence of myocardial necrosis in a clinical setting onsistent with acute myocardial ischemia. Under this conditıon one of the following critera meet the diagnosis of Mİ: Detectıon of a rise an/or fall of cardiac biomarkers (cTn) with at least one value above 99th percantile of URL and with at least one of the following: -+ ST-T changes or new LBB. -+ Development pathological Q wave . -+İmaqging new loss of viable myocardium or regional wall motıon abnormality. -+ İdentificatıon of intracoronary thrombus by angiography.")
29
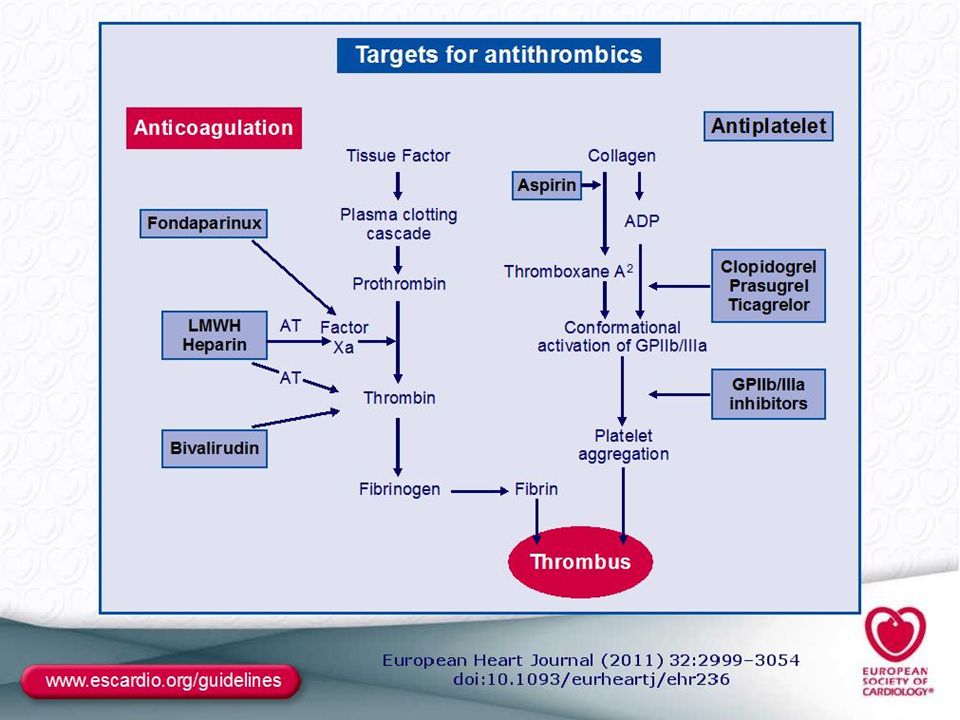
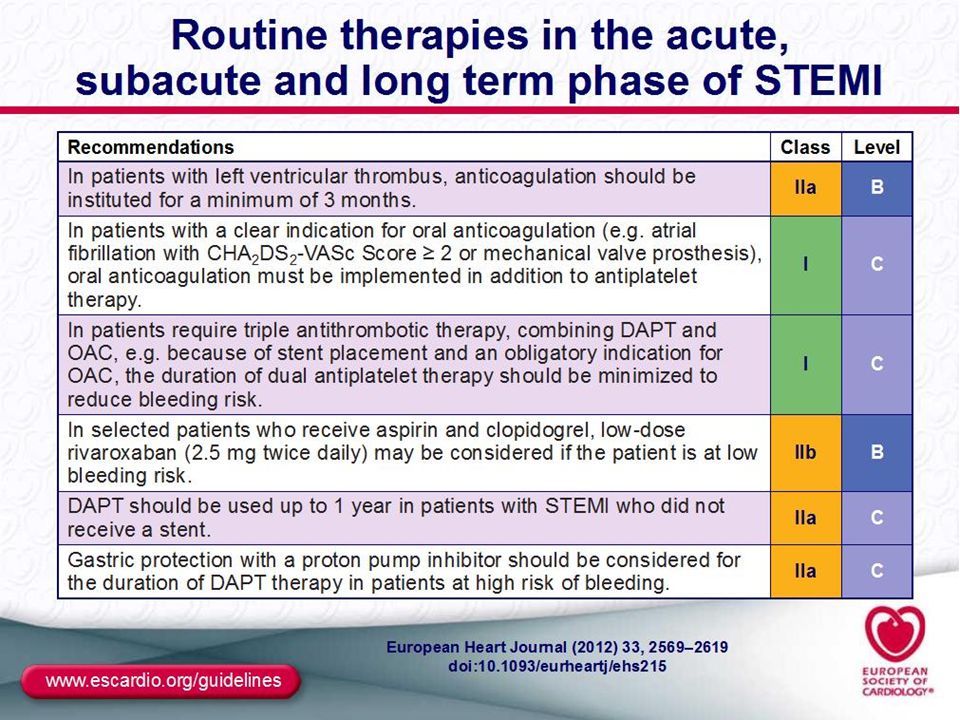
MANAGEMENT OF STEMİ: The aim of therapy: Reperfusion therapy (RPT) is indicated in all patients with symptoms of <12 h duration and persistent ST-segment elevation or (presumed) new LBBB. Primary principal of RPT: Reperfusıon (open occluded artery)should be performed as soon as possible anwith maintain patency of open artery at least 1year. 2 Typies of RP therapies are indicated; Percutaneous interventıon (PCI) and Fibrinolytic Therapy (FLT). Time-to- threatment ( delay of RP) is the key of sucess. When RP perform in patient within 1h of symptom onset, Mİ could be aborted . Rule of Reperfusıon : RPT Should be introduced “timely” to all patient with persistent STE within 12h of symptom onset. Timely RP (delay of therapy): Time from presentatıon in First medical contact (FMC), or Hospital emergency room (door) to -needle (FLT) or/ -baloon inflatıon (PCI). Recomended delays: Door- needle: <30 minutes and door- balon <90 min (PCI capable Hxt) or <120 min (Non-PCI capable Hxt).Patient presenting early with large Mİ PCI delay should be <60min. Adjunctive antithrombotic therapy to RP therapies : Antitrombotics therapy is the cornerstone of RPT to establısh acute reperfusıon and maintain sustain patency. FLT: with concurrently; aspirin, Clopidegrol and Low Molecular weight Heparin (Enoxaparin). PCI : At the table; Aspirin Prasugrel/clopidogrel, Bivaluridin or heparins.
 is indicated in all patients with symptoms of <12 h duration and persistent ST-segment elevation or (presumed) new LBBB. Primary principal of RPT: Reperfusıon (open occluded artery)should be performed as soon as possible anwith maintain patency of open artery at least 1year. 2 Typies of RP therapies are indicated; Percutaneous interventıon (PCI) and Fibrinolytic Therapy (FLT). Time-to- threatment ( delay of RP) is the key of sucess. When RP perform in patient within 1h of symptom onset, Mİ could be aborted . Rule of Reperfusıon : RPT Should be introduced timely to all patient with persistent STE within 12h of symptom onset. Timely RP (delay of therapy): Time from presentatıon in First medical contact (FMC), or Hospital emergency room (door) to -needle (FLT) or/ -baloon inflatıon (PCI). Recomended delays: Door- needle: <30 minutes and door- balon <90 min (PCI capable Hxt) or <120 min (Non-PCI capable Hxt).Patient presenting early with large Mİ PCI delay should be <60min. Adjunctive antithrombotic therapy to RP therapies : Antitrombotics therapy is the cornerstone of RPT to establısh acute reperfusıon and maintain sustain patency. FLT: with concurrently; aspirin, Clopidegrol and Low Molecular weight Heparin (Enoxaparin). PCI : At the table; Aspirin Prasugrel/clopidogrel, Bivaluridin or heparins.")
30
Summary of important delays and treatment goals in the management of acute ST-segment elevation myocardial infarction: FMC- ECG and Diagnosıs: ≤ 10 min. PCI- Hospital: Door-to baloon: ≤90 min. (Early presentatıon with large Mİ): ≤60 min). Not-PCI-Hospital: FMC-Baloon: ≤120 min. FLT: Door (FMC)- Needle: ≤30 min. SELECTION OF RP THERAPİES : Primary PCI is the recommended reperfusion therapy over fibrinolysis, if performed by an experienced team within120 min of FMC. Primary PCI is indicated for patients with severe acute heart failure or cardiogenic shock, unless the expected PCI related delay is not excessive and the patient presents early after symptom onset. FLT is recommended within 12 h of symptom onset in patients without contraindications, if primary PCI cannot be performed by an experienced team within 120 min of FMC.Fibrin-specific fibrinolytics prefered (Alteplase, Tenecteplase PA)
: ≤60 min). Not-PCI-Hospital: FMC-Baloon: ≤120 min. FLT: Door (FMC)- Needle: ≤30 min. SELECTION OF RP THERAPİES : Primary PCI is the recommended reperfusion therapy over fibrinolysis, if performed by an experienced team within120 min of FMC. Primary PCI is indicated for patients with severe acute heart failure or cardiogenic shock, unless the expected PCI related delay is not excessive and the patient presents early after symptom onset. FLT is recommended within 12 h of symptom onset in patients without contraindications, if primary PCI cannot be performed by an experienced team within 120 min of FMC.Fibrin-specific fibrinolytics prefered (Alteplase, Tenecteplase PA)")
31
İNDİCATIONS AND STRATEGY OF REPERFUSION THERAPY:
1- Reperfusion therapy is indicated in all patients present with symptoms of <12 h duration and persistent ST-segment elevation or (presumed) new LBBB. Prefered RPT is PPCI (if performed timely). FLT is alternative of PPCI. 2- If FMC is EMS or at a non-PCI-capable centre, patient immediately transfer to the catheterization laboratory for PCI. 3- In settings where primary PCI cannot be performed within 120 min of FMC by an experienced team, fibrinolysis should be considered, particularly if it can be given pre-hospital (e.g. in the ambulance) and within the first 120 min of symptom onset . After administratıon of FLT, patients should be transfer to PCI-center for early anggıography. Primary PCI (— defined as an emergent percutaneous catheter intervention in the setting of STEMI, without previous fibrinolytic treatment —) is the preferred reperfusion strategy in patients with STEMI; when provided it can be performed rapidly (i.e. within guideline-mandated times), by an experienced team, regardless of whether the patient presents to a PCI-capable hospital.
 new LBBB. Prefered RPT is PPCI (if performed timely). FLT is alternative of PPCI. 2- If FMC is EMS or at a non-PCI-capable centre, patient immediately transfer to the catheterization laboratory for PCI. 3- In settings where primary PCI cannot be performed within 120 min of FMC by an experienced team, fibrinolysis should be considered, particularly if it can be given pre-hospital (e.g. in the ambulance) and within the first 120 min of symptom onset . After administratıon of FLT, patients should be transfer to PCI-center for early anggıography. Primary PCI (— defined as an emergent percutaneous catheter intervention in the setting of STEMI, without previous fibrinolytic treatment —) is the preferred reperfusion strategy in patients with STEMI; when provided it can be performed rapidly (i.e. within guideline-mandated times), by an experienced team, regardless of whether the patient presents to a PCI-capable hospital.")
32
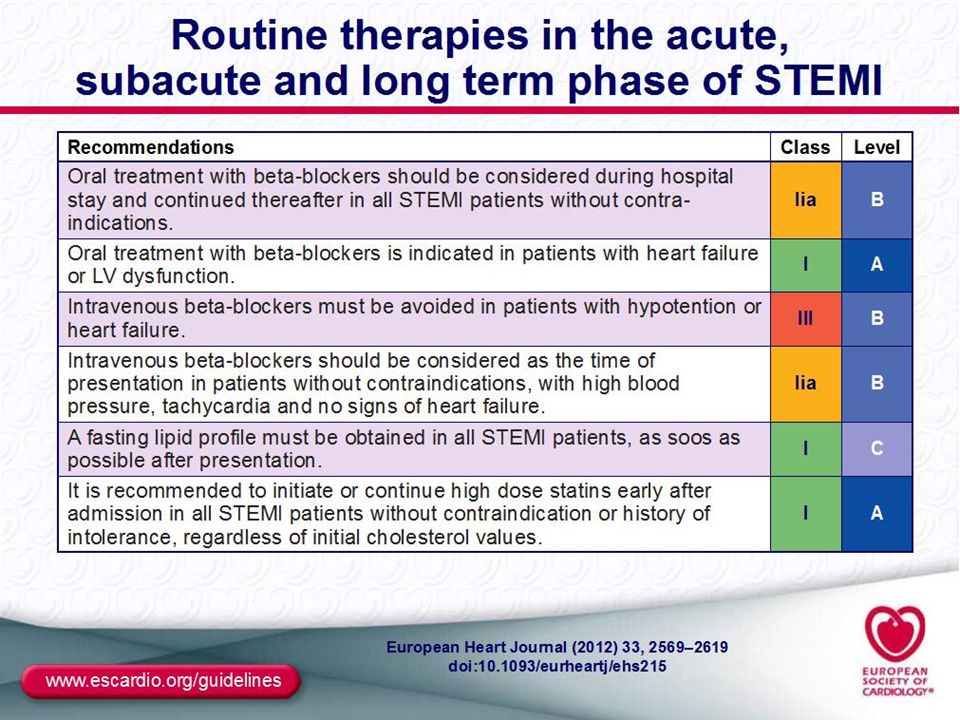
İnitial therapy: Aspirin, should be diagnosed with given as soon as posible to patient with symptoms of high likelihood ACS. Clopidegrol, should be combined to ASA after STEMİ diagnosed by ECG and rapidly evaluate for Emergent RP strategy . Nitrates, sublingual Nitroglycerin for testing ischaemia or relieve for chest discomfort. Oxygen, with nasal canul is traditional aproach, high concantratıon O2 inhalatıon should be avoided in for COPD patients . Morphin, use for control anxiety in patients unresponsive to nitrates therapy. Therapies in first 24h: Oral Beta bloker: Oral should be given all patients unless has symptoms and sign of heart failure, complete heart block and severe bradycardia. ACEİ: Should be given partıcularly nonreperfused, unseccesful RP and large Mİ with systolic LV dysfunctıon. Statin regardless the cholesterol levels. Aldasteron antagonist: İn patients dysfunctıon on ACEİ and beta blocker therapy with large Mİ and systolic LV dysfunctıonshould be combined untill hyperkalemia and moderate renal dyfunctıon. Discharged therapies: These drugs should be included to patient’s therapy: ASA, Clopidegrol ( for PCI Prasugrel), BB, ACEİ, aldosteron antagonist ( given in first 2 weeks) and statin.
, BB, ACEİ, aldosteron antagonist ( given in first 2 weeks) and statin.")
33
NON ST ELEVATION ACS (NSTE):
Patients with present with ischaemic chest pain >20 minutes duratıon, with ECG changies rather than persistent STE ; diagnosed is NSTE- ACS. Biomarkers (troponins) further distinguish NSTEMI and unstable angina. Its Pathological, imaging have demonstrated atherosclerotic plaque rupture or erosion, with differing degrees of superimposed platelet thrombosis and vasospasm with non-total lumen occlusıon and distal embolization. When compared STEMİ, because of less severe thrombogenic burden but more sustain prothrombotic activity, this patients has higher rate of subsequent late ischemic events risk (recurrent ischemia, Mİ and ischemia required rehospitalisatıon ) even lower inhospital mortality and morbidity . Clinical presentation The clinical presentation of NSTE-ACS encompasses a wide variety of symptoms. Traditionally, several clinical presentations have been distinguished: † Prolonged (.20 min) anginal pain at rest. † New onset (de novo) angina (Class II or III of the Classification of the CCS. † Recent destabilization of previously stable angina with at least Canadian CCS Class III angina characteristics. (crescendo angina); or † Post-MI angina.
 further distinguish NSTEMI and unstable angina. Its Pathological, imaging have demonstrated atherosclerotic plaque rupture or erosion, with differing degrees of superimposed platelet thrombosis and vasospasm with non-total lumen occlusıon and distal embolization. When compared STEMİ, because of less severe thrombogenic burden but more sustain prothrombotic activity, this patients has higher rate of subsequent late ischemic events risk (recurrent ischemia, Mİ and ischemia required rehospitalisatıon ) even lower inhospital mortality and morbidity . Clinical presentation. The clinical presentation of NSTE-ACS encompasses a wide variety of symptoms. Traditionally, several clinical presentations have been distinguished: † Prolonged (.20 min) anginal pain at rest. † New onset (de novo) angina (Class II or III of the Classification of the CCS. † Recent destabilization of previously stable angina with at least Canadian CCS Class III angina characteristics. (crescendo angina); or. † Post-MI angina.")
34
MANAGEMENT STRATEGY: In patients with a suspected NSTE-ACS, diagnosis and short-term ischaemic and bleeding risks stratification should be evaluate based on a combination of clinical history, symptoms, physical findings, ECG (repeated or continuous ST monitoring), and biomarkers. 12-lead ECG should be obtained within 10 min after first medical contact and immediately read. NSTE- ACS patients should be admitted preferably to chest pain units or coronary care units. It is recommended to use established risk scores for prognosis and bleeding (e.g. TIMI, GRACE and CRUSADE risk score systems). Blood has to be drawn promptly for troponin (cardiac troponin T or I) measurement. The result should be available within 60 min. The test should be repeated 6–9 h after initial assessment if the first measurement is not conclusive. Initial therapeutic measures: Aspirin, Oxygen, Nitrates, Morphine (!). Treatments when an NSTE- ACS diagnosis appears likely: P2Y12 inhibitor (Ticagrelor or Clopidegrol) and Anticoagulatıon (Fondaparinux or Enoxaparin) should be combined Aspirin (Preferably inhospital admısıon) and oral Beta blocker given in 24h of hospital admision if not contraindicated . Ticagrelor for high risk NSTE- ACS patiens proceed to PCI. Because high frequency of reccurence ischaemic event rate; İnvasive strategy is the mainstay of therapy, NSTE- ACS patient with İntermediate and high risk should referred to invasive strategy inhospital course.
, and biomarkers. 12-lead ECG should be obtained within 10 min after first medical contact and immediately read. NSTE- ACS patients should be admitted preferably to chest pain units or coronary care units. It is recommended to use established risk scores for prognosis and bleeding (e.g. TIMI, GRACE and CRUSADE risk score systems). Blood has to be drawn promptly for troponin (cardiac troponin T or I) measurement. The result should be available within 60 min. The test should be repeated 6–9 h after initial assessment if the first measurement is not conclusive. Initial therapeutic measures: Aspirin, Oxygen, Nitrates, Morphine (!). Treatments when an NSTE- ACS diagnosis appears likely: P2Y12 inhibitor (Ticagrelor or Clopidegrol) and Anticoagulatıon (Fondaparinux or Enoxaparin) should be combined Aspirin (Preferably inhospital admısıon) and oral Beta blocker given in 24h of hospital admision if not contraindicated . Ticagrelor for high risk NSTE- ACS patiens proceed to PCI. Because high frequency of reccurence ischaemic event rate; İnvasive strategy is the mainstay of therapy, NSTE- ACS patient with İntermediate and high risk should referred to invasive strategy inhospital course.")
35
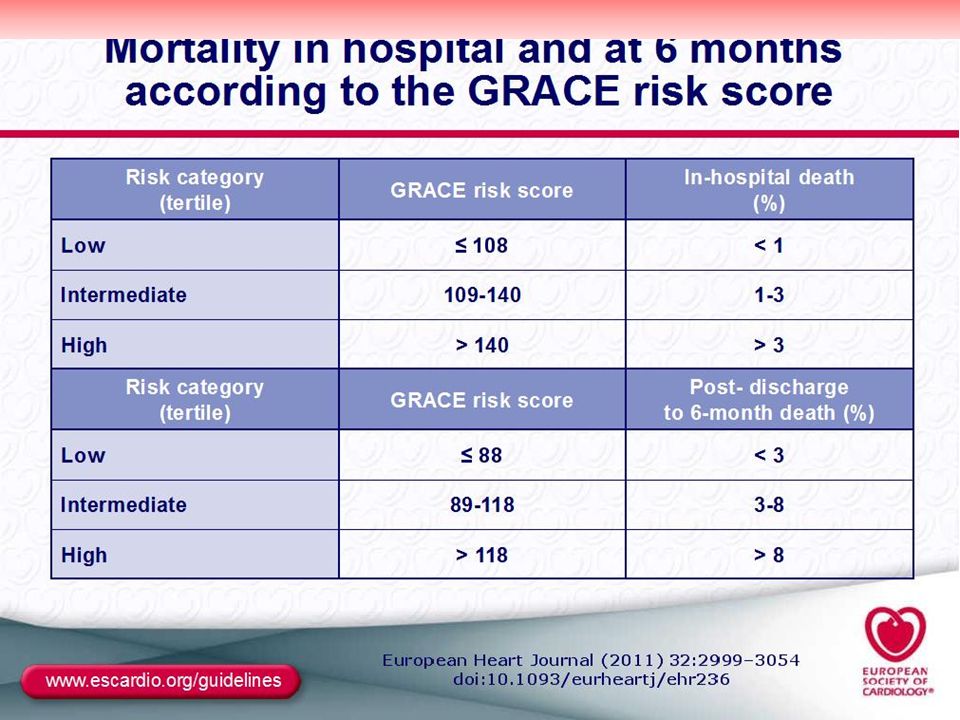
TIMI Risk score: GRACE Risk score –
İndicators of 35 days composite events risks (Mortality, or new, reMİ, or severe recurrent ischemia reguiring urgent revascularizastıon through 14 day after admision). • Age ≥65 years At least 3 risk factors, • Prior coronary stenosis ≥50% • ST- segment deviation on ECG presentatıon, • At least >2 anginal events in the proceeding 24 hours, • Aspirin treatment in the prior 7 days, • Increased cardiac biomarkers, • Prior congestive heart failure, MI, CABG or PCI. Each characteristic qualifies for one point in the risk score 0-1: %4.7; 3: %%13.2; 6-7: %40.9. GRACE Risk score – - (All cause mortality risk in hospital and at 6 months). • Killip class (heart failure) • Arterial blood pressure • ST deviation on ECG • Cardiac arrest • Increased creatinine concentration • Increased CK-MB or troponin concentration • Heart rate. Risk score: >140- (İn hospital death >%3); Risk score >118: - (at 6 month death %8).
. • Age ≥65 years. At least 3 risk factors, • Prior coronary stenosis ≥50% • ST- segment deviation on ECG presentatıon, • At least >2 anginal events in the proceeding 24 hours, • Aspirin treatment in the prior 7 days, • Increased cardiac biomarkers, • Prior congestive heart failure, MI, CABG or PCI. Each characteristic qualifies for one point in the risk score. 0-1: %4.7; 3: %%13.2; 6-7: %40.9. GRACE Risk score – - (All cause mortality risk in hospital and at 6 months). • Killip class (heart failure) • Arterial blood pressure. • ST deviation on ECG. • Cardiac arrest. • Increased creatinine concentration. • Increased CK-MB or troponin concentration. • Heart rate. Risk score: >140- (İn hospital death >%3); Risk score >118: - (at 6 month death %8).")
36
Risk Calculator for 6-Month Postdischarge Mortality After Hospitalization for Acute Coronary Syndrome Record the points for each variable at the bottom left and sum the points to calculate the total risk score

38
İNVASİVE STRATEGY: Cardiac catheterization followed by revascularization has been shown to prevent recurrent ischaemia and/or improve short and long-term outcomes. İnvasive approach in higher risk NSTE- ACS patients showed approximately %20-30 reductıon risk of 2 years mortality, non-fatal Mİ and reccurent USAP. Several risk factors (troponin elevation, diabetes, ST-segment depression, renal insufficiency, etc.) have been identified to predict the long-term benefit of an invasive strategy. Depending on the acuteness of risk, the timing of angiography can be tailored, according to four categories: _ invasive (,72 h); – urgent invasive (,120 min); – early invasive (,24 h); _ primarily conservative. The optimal timing depends on the risk profile of the individual patient and can be assessed by several variables
 have been identified to predict the long-term benefit of an invasive strategy. Depending on the acuteness of risk, the timing of angiography can be tailored, according to four categories: _ invasive (,72 h); – urgent invasive (,120 min); – early invasive (,24 h); _ primarily conservative. The optimal timing depends on the risk profile of the individual patient and can be assessed by several variables")
39
Urgent invasive strategy (<120 min after first medical contact):
This should be undertaken for very high risk patients. These patients are characterized by: - Refractory angina (indicating evolving MI without ST abnormalities). - Recurrent angina despite intense antianginal treatment, associated with ST depression (2 mm) or deep negative T waves. - Clinical symptoms of heart failure or haemodynamic instability (‘shock’). - Life-threatening arrhythmias (ventricular fibrillation or ventricular tachycardia). Early invasive strategy (<24 h after first medical contact) Most patients initially respond to the antianginal treatment, but are at increased risk and need angiography followed by revascularization: - High risk patients as identified by a GRACE risk score >140 and/or the presence of at least one (following) primary high risk criterion should undergo invasive evaluation within 24 h. Relevant rise and /or fall cTn. Dynamic ST-Twave changes(silent or symptomatic).
. - Recurrent angina despite intense antianginal treatment, associated with ST depression (2 mm) or deep negative T waves. - Clinical symptoms of heart failure or haemodynamic instability (‘shock’). - Life-threatening arrhythmias (ventricular fibrillation or ventricular. tachycardia). Early invasive strategy (<24 h after first medical contact) Most patients initially respond to the antianginal treatment, but are at increased risk and need angiography followed by revascularization: - High risk patients as identified by a GRACE risk score >140 and/or the presence of at least one (following) primary high risk criterion should undergo invasive evaluation within 24 h. Relevant rise and /or fall cTn. Dynamic ST-Twave changes(silent or symptomatic).")
40
Invasive strategy (<72 h after first medical contact)
In patients with less acute risk and without recurrence of symptoms, angiography may be performed within a time window of 72 h. Thus, such patients should undergo elective invasive evaluation at the first opportunity depending on the local circumstances. Conservative strategy (no or elective angiography) Patients that fulfil all of the following criteria may be regarded as low risk and should not routinely be submitted to early invasive evaluation: † No recurrence of chest pain. † No signs of heart failure. † No abnormalities in the initial ECG or a second ECG (at 6–9 h). † No rise in troponin level (at arrival and at 6–9 h). † No inducible ischaemia. Therapy: ASA, Clopidegrol, Enoxaparin, Beta blocker, Statin.
 Patients that fulfil all of the following criteria may be regarded as low risk and should not routinely be submitted to early invasive evaluation: † No recurrence of chest pain. † No signs of heart failure. † No abnormalities in the initial ECG or a second ECG (at 6–9 h). † No rise in troponin level (at arrival and at 6–9 h). † No inducible ischaemia. Therapy: ASA, Clopidegrol, Enoxaparin, Beta blocker, Statin.")
41
STABLE ANGİNA: Stable angina is defined as short duration chest and/or arm discomfort that shows no change in the past 60 days in frequency, duration, or precipitating causes. Most often pain duration is less than 10 minutes, and rarely up to 15 minutes; the mild-to-moderate discomfort is relieved within 1 to 10 minutes by cessation of the precipitating activity or use of sublingual nitroglycerin. In more than 90% of patients, stable angina is caused by a greater than 70% obstruction in at least one coronary artery. In less than 10% of patient wit5h lesser degree of atheromatous obstruction, coronary artery spasm, or small vessel disease is present.

42
Pathophysiology: Myocardial ischaemia is a dynamic process. It is now clear that three, not two, determinants play a major role in the pathogenesis of myocardial ischaemia, which may manifest as the chest pain of angina or remain painless ( termed silent ischaemia). The three determinants of myocardial ischaemia are as follows: •1. Concentric or eccentric coronary atheroma causing greater than about 70% stenosis. Concentric plaques are observed mainly with stable angina and there is a tendency for them to be eccentric in patients with frequent rest pain and in those with unstable angina. • 2. Increased myocardial oxygen demand. • 3. Release of catecholamines occurring at the onset of angina and during the episode in most patients with stable angina. Release of catecholamines may actually initiate ischemia, which stimulates further catecholamine release, and a vicious circle perpetuates the oxygen lack
. The three determinants of myocardial ischaemia are as follows: •1. Concentric or eccentric coronary atheroma causing greater than about 70% stenosis. Concentric plaques are observed mainly with stable angina and there is a tendency for them to be eccentric in patients with frequent rest pain and in those with unstable angina. • 2. Increased myocardial oxygen demand. • 3. Release of catecholamines occurring at the onset of angina and during the episode in most patients with stable angina. Release of catecholamines may actually initiate ischemia, which stimulates further catecholamine release, and a vicious circle perpetuates the oxygen lack.")
43
The major Symptoms of chronic ischaemic heart disease is angina pectoris, with a clinical diagnosis based on five features: 1) The character of pain is deep visceral pressure or squeezing sensatıon, rather than sharp or stabbing orpinbrick-like pain. İt is quality deep visceral and intense it make the patients pay atentıon, but not excruciating. Area of pain not located and precipitated with 2 fingertips 2) The pain almost always has some substernal component, although some patient complain of pain only on the right or left side of the chest, upper back, or epigastrium. 3) The pain may radiate from the thorax to the jaw, neck or arm (usually ulnar surface of left arm). 4) Angina usually precipitated exertıon, emotional upset, or other events that increased myocardial oxygen demand (tachyarrhythmias, extreme elevatıons in blood pressure). 5) Angina pectoris is transient, lasting between minutes.It is relieved (within 1- 5 minutes) by sessatıon of the precipitating events (exercise) or taking sublingual nitroglycerin (within 1-2 minutes). Chest pain that last longer than 30 minutes it is more consistent with Mİ pain; pain < 2 minutes is unlikely due to myocardial ischaemia.
 The character of pain is deep visceral pressure or squeezing sensatıon, rather than sharp or stabbing orpinbrick-like pain. İt is quality deep visceral and intense it make the patients pay atentıon, but not excruciating. Area of pain not located and precipitated with 2 fingertips. 2) The pain almost always has some substernal component, although some patient complain of pain only on the right or left side of the chest, upper back, or epigastrium. 3) The pain may radiate from the thorax to the jaw, neck or arm (usually ulnar surface of left arm). 4) Angina usually precipitated exertıon, emotional upset, or other events that increased myocardial oxygen demand (tachyarrhythmias, extreme elevatıons in blood pressure). 5) Angina pectoris is transient, lasting between minutes.It is relieved (within 1- 5 minutes) by sessatıon of the precipitating events (exercise) or taking sublingual nitroglycerin (within 1-2 minutes). Chest pain that last longer than 30 minutes it is more consistent with Mİ pain; pain < 2 minutes is unlikely due to myocardial ischaemia.")
44
Diagnosis is based on a careful relevant history.
Distinctive characteristics: The pain of angina has certain distinctive characteristics; Aaretrosternal discomfort precipitated by a particular activity, especially walking quickly up an incline or against the wind. • The discomfort is a tightness, constriction, squeezing, heaviness, pressure, strangulation, burning, nausea, or an indigestion-like feeling of gradual onset that disappears at rest. Occasionally, the pain is described as sharp, and at times discomfort is replaced by shortness of breath on exertion. • The area of pain is usually at least the size of a clenched fist, often occupying most of the central chest area. The patient uses two or more fingers, the entire palm of the hand, or the fist to indicate the pain site. A finger or pencil point area of pain is rarely caused by myocardial ischaemia. •Relief of pain in an individual with stable angina always occurs within minutes of cessation of the precipitating exertional or emotional activity. Relief with sublingual nitroglycerin occurs promptly within 1–2 minutes. Pain or discomfort disappears within 1 to 5 minutes of stopping the precipitating activity. Discomfort may start in the lower, middle, or upper substernal area, the lower jaw, or the arm

45
The Canadian Cardiovascular Society (CCS) grading of angina is widely used to differentiate mild, moderate, or severe stable angina: • Class 1 Angina: Pain is precipitated only by severe and usually prolonged exertion. • Class 2 Angina: Pain on moderate effort, for example, precipitated by walking uphill or by walking briskly for more than three blocks on the level in the cold, against a wind, or provoked by emotional stress. There is “slight limitation of ordinary activity.” • Class 3 Angina: Marked limitation of ordinary activity; pain occurs on mild exertion, *usually restricting daily chores. Unable to walk two blocks on the level at comfortable temperatures and at a normal pace. • Class 4 Angina: Chest discomfort on almost any physical activity, for example, dressing, shaving, walking less than 100 feet indoors. Pain may be present at rest.

46
THERAPY: Patients suitable for medical management usually have two of the following characteristics: • Stable, functional class 1 or 2 angina. • Good effort tolerance, negative or weakly positive treadmill exercise test . Patients who are unable to exercise because of intermittent claudication or arthritis cannot be graded as class 1 or 2. • Good ventricular function( EF), or estimate greater than 50%. • Absence of left main coronary artery disease. • Presence of double vessel disease in the absence of severe proximal stenosis of the LAD artery with normal EF. • Concomitant comorbid disease and contraindications to bypass surgery. • Age over 80 and not in good general health. • Lesions not ideal for intervention. ►Most patients with stable class 1 and 2 angina are managed with sublingual nitroglycerin and a one-a-day β-blocker plus aspirin and a statin to keep the LDL less than 2 mmol/L (80 mg/dL), for targeted eblood pressure <135/85 mmHg with calcıum antagonist, ACEİ/ARB or combinatıons. Duidlines recomended d secondary preventıon measures should be implemented .
, or estimate greater than 50%. • Absence of left main coronary artery disease. • Presence of double vessel disease in the absence of severe proximal stenosis of the LAD artery with normal EF. • Concomitant comorbid disease and contraindications to bypass surgery. • Age over 80 and not in good general health. • Lesions not ideal for intervention. ►Most patients with stable class 1 and 2 angina are managed with sublingual nitroglycerin and a one-a-day β-blocker plus aspirin and a statin to keep the LDL less than 2 mmol/L (80 mg/dL), for targeted eblood pressure <135/85 mmHg with calcıum antagonist, ACEİ/ARB or combinatıons. Duidlines recomended d secondary preventıon measures should be implemented .")
47
Q’s of the İHD for Medical students:??.
1- Earliest characteristics of atherosclerosıs?. Fatty streaks endotelial dysfunctıon, Lipid accumulatıon, Thrombosıs. 2- Sıgn of Subclinical atherosclerosıs? a) Fatty streak b) fibrous plaque with <%50 luminal diameter narrowing. c) Macroscopic Normal coronary arteries. 3- Mechanism of stable angina? Decreased coronary flow with decreased oxygen demand İncreased myocardial oxygen demand not matching augmented coronary flow, Acute plaque disruptıon Vazospasm. 4- Modifiable CAD risk factors? a) Age, genetics, gender b) tobacco smoking, diabetes, hypertensıon ECG changes, partıcularly Q wave. 5- What is the best Primary therapy for these ischemic syndromes ?. Syndromes: a) NSTE ACS b) STEMİ, stable angina Selected Therapy: i) Fibrinolytics; ii) Primary PCI; iii) Nitroglycerin and beta blocker;i) combinatıon of antiplatelet and anticoagulan drugs. 6- What is your fırst order?: Patient presentation with ishemic symptoms of <6 h duratıon and ST elevatıon on ECG. a) Measured troponin level emergently; b) give Aspirin; c) referred to the general hospital; d) Give sublingual nitrogliserin; e) Transport to the PCI-center if ( minutes) or give (---) in (----minutes).
 Fatty streak. b) fibrous plaque with <%50 luminal diameter narrowing. c) Macroscopic Normal coronary arteries. 3- Mechanism of stable angina Decreased coronary flow with decreased oxygen demand. İncreased myocardial oxygen demand not matching augmented coronary flow, Acute plaque disruptıon. Vazospasm. 4- Modifiable CAD risk factors a) Age, genetics, gender. b) tobacco smoking, diabetes, hypertensıon. ECG changes, partıcularly Q wave. 5- What is the best Primary therapy for these ischemic syndromes . Syndromes: a) NSTE ACS b) STEMİ, stable angina. Selected Therapy: i) Fibrinolytics; ii) Primary PCI; iii) Nitroglycerin and beta blocker;i) combinatıon of antiplatelet and anticoagulan drugs. 6- What is your fırst order : Patient presentation with ishemic symptoms of <6 h duratıon and ST elevatıon on ECG. a) Measured troponin level emergently; b) give Aspirin; c) referred to the general hospital; d) Give sublingual nitrogliserin; e) Transport to the PCI-center if ( minutes) or give (---) in (----minutes).")
49
2- Inflammatory cell recruitment:
1) Endothelial dysfunction in atherosclerosis is accompanied by endothelial cell activation, which results in expression of cell surface adhesion molecules (the selectins and VCAM-1). 2) These molecules mediate recruitment of inflammatory cells (monocytes and T cells). Inflammatory cell accumulation is also increased by smooth muscle cells and accumulation of modified lipid (e.g. oxidized low-density lipoprotein, LDL). • Local rheological factor effects (differences in blood flow, shear stress and endothelial cell biology ) determine side of the vascular susceptibilty. 3) Monocytes recruited into the vessel wall ingest modified lipids and become activated foam cells. Necrotic foam cells contribute to the lipid core of developing plaques. In addition, activated macrophages and T cells express pro-inflammatory cytokines and growth factors, which maintain inflammatory cell recruitment and stimulate endothelial activation and smooth muscle cell proliferation. 3- Vascular smooth muscle cells Vascular smooth muscle cells in the vascular media are normally quiescent and contractile. 4) In response to vascular injury, cytokines and growth factors (e.g interleukins, interferon-γ, tumour necrosis factor α), smooth muscle cells can change to an activated phenotype that allows them to migrate, proliferate and synthesize extracellular matrix proteins (collagen and elastin). These factors contribute to the growth of the plaque. 5) Smooth muscle cell migration and proliferation with extracellular matrix synthesis may contribute to luminal narrowing. However, formation of a strong fibrous cap by smooth muscle cells, is important in maintaining plaque stability by isolating the lipid core and inflammatory cells from circulating blood
 Endothelial dysfunction in atherosclerosis is accompanied by endothelial cell activation, which results in expression of cell surface adhesion molecules (the selectins and VCAM-1). 2) These molecules mediate recruitment of inflammatory cells (monocytes and T cells). Inflammatory cell accumulation is also increased by smooth muscle cells and accumulation of modified lipid (e.g. oxidized low-density lipoprotein, LDL). • Local rheological factor effects (differences in blood flow, shear stress and endothelial cell biology ) determine side of the vascular susceptibilty. 3) Monocytes recruited into the vessel wall ingest modified lipids and become activated foam cells. Necrotic foam cells contribute to the lipid core of developing plaques. In addition, activated macrophages and T cells express pro-inflammatory cytokines and growth factors, which maintain inflammatory cell recruitment and stimulate endothelial activation and smooth muscle cell proliferation. 3- Vascular smooth muscle cells. Vascular smooth muscle cells in the vascular media are normally quiescent and contractile. 4) In response to vascular injury, cytokines and growth factors (e.g interleukins, interferon-γ, tumour necrosis factor α), smooth muscle cells can change to an activated phenotype that allows them to migrate, proliferate and synthesize extracellular matrix proteins (collagen and elastin). These factors contribute to the growth of the plaque. 5) Smooth muscle cell migration and proliferation with extracellular matrix synthesis may contribute to luminal narrowing. However, formation of a strong fibrous cap by smooth muscle cells, is important in maintaining plaque stability by isolating the lipid core and inflammatory cells from circulating blood.")
56
Atherosclerosis is a disease of the arterial wall that underlies many of the common causes of cardiovascular morbidity and mortality, including myocardial infarction (MI), peripheral vascular disease and cerebrovascular disease. PLAQUE Morphology/Structure • Plaque cap thickness • Plaque lipid core size • Plaque stenosis (luminal narrowing) • Remodeling (expansive vs constrictive remodeling) • Color (yellow, glistening yellow, red, etc) • Collagen content versus lipid content, mechanical stability (stiffness and elasticity) • Calcification burden and pattern (nodule vs scattered, superficial vs deep, etc) • Shear stress (flow pattern throughout the coronary artery The risk of a vulnerable patient is affected by vulnerable plaque and/or vulnerable blood and/or vulnerable myocardium. A comprehensive assessment must consider all of the above.
 • Remodeling (expansive vs constrictive remodeling) • Color (yellow, glistening yellow, red, etc) • Collagen content versus lipid content, mechanical stability (stiffness and elasticity) • Calcification burden and pattern (nodule vs scattered, superficial vs deep, etc) • Shear stress (flow pattern throughout the coronary artery The risk of a vulnerable patient is affected by vulnerable plaque and/or vulnerable blood and/or vulnerable myocardium. A comprehensive assessment must consider all of the. above.")
58
Correlation between frequencyof plaques, degree of stenosis, and riskof complication per plaque as a functionof plaque progression. Although theaverage absolute risk of severely stenoticplaques may be higher than the averageabsolute risk of mildly stenotic plaques,there are more plaques with mild stenosesthan plaques with severe stenoses.

59
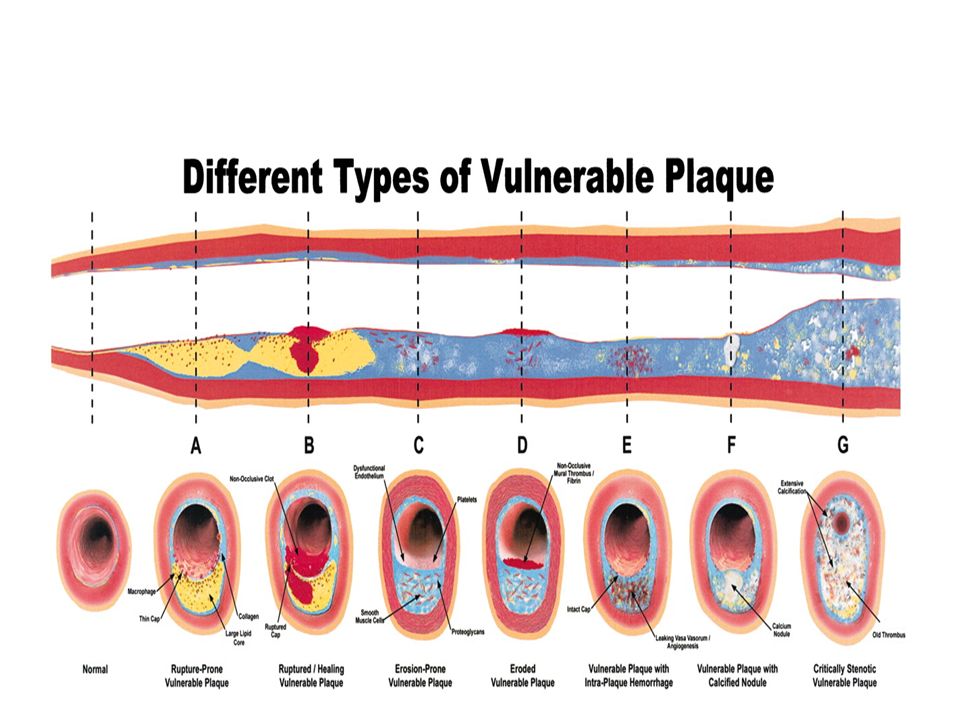
What Is a Vulnerable Plaque
What Is a Vulnerable Plaque? In the upper panel, the middle figure shows a presumed vulnerable plaque, a thin-capped atheroma with a large lipid/necrotic core and a thin fibrous cap infiltrated by inflammatory cells, which is thought to be the immediate precursor of symptomatic thrombosed plaque (upper right figure). in the lower panel, a “vulnerable plaque” The true precursor to a symptomatic thrombosed plaque might depend on such factors as the exact cap thickness, size of the lipid/necrotic core, inflammatory cell volume, thrombogenicity of the blood, and so forth. .
. in the lower panel, a vulnerable plaque The true precursor to a symptomatic thrombosed plaque might depend on such factors as the exact cap thickness, size of the lipid/necrotic core, inflammatory cell volume, thrombogenicity of the blood, and so forth. .")
60
Microanatomy of coronary arterial thrombosis and acute occlusion

61
Clinicopathologic correlation of asymptomatic atherosclerosis leading to symptomatic atherothrombosis.

62
PLAQUE PROGRESSION CLASİFİCATION (Stary)
Phase 1 (early) Lesions are small, commonly seen in young people, and categorized into three types as follows: type I lesions, consisting of macrophage-derived foam cells that contain lipid droplets; type II lesions, consisting of both macrophages and smooth-muscle cells and mild extracellular lipid deposits; and type III lesions, consisting of smooth-muscle cells surrounded by extracellular connective tissue, fibrils, and lipid deposits. Phase 2 (advanced) Lesions, although not necessarily stenotic, may be prone to rupture because of their high lipid content, increased inflammation, and thin fibrous cap. These plaques are categorized morphologically as one of two variants: type IV lesions, consisting of confluent cellular lesions with a great deal of extracellular lipid intermixed with normal intima, which may predominate as an outer layer or cap; or type Va lesions, possessing an extracellular lipid core covered by an acquired fibrous cap. Phase 2 plaques can evolve into the acute phases 3 and 4. Phase 3 These lesions are characterized by acute complicated type VI lesions, originating from ruptured (type IV or Va) or eroded lesions, and leading to mural, non-obstructive thrombosis. This process is clinically silent, but occasionally may lead to the onset of angina . Phase 4 These lesions are characterized by acute complicated type VI lesions, with fixed or repetitive occlusive thrombosis. This process becomes clinically apparent in the form of an acute coronary syndrome (ACS), although not infrequently it is silent . About two-thirds of ACS are caused by occlusive thrombosis on a non-stenotic plaque, although in about one-third, the thrombus occurs on the surface of a stenotic plaque . In phases 3 and 4, changes in the geometry of ruptured plaques, as well as organization of the occlusive or mural thrombus by connective tissue, can lead to the occlusive or significantly stenotic and fibrotic plaques. Phase 5 These lesions are characterized by type Vb (calcific) or Vc (fibrotic) lesions that may cause angina; however, if preceded by stenosis or occlusion with associated ischemia, the myocardium may be protected by collateral circulation and such lesions may then be silent or clinically inapparent.
 Lesions are small, commonly seen in young people, and categorized into three types as follows: type I lesions, consisting of macrophage-derived foam cells that contain lipid droplets; type II lesions, consisting of both macrophages and smooth-muscle cells and mild extracellular lipid deposits; and type III lesions, consisting of smooth-muscle cells surrounded by extracellular connective tissue, fibrils, and lipid deposits. Phase 2 (advanced) Lesions, although not necessarily stenotic, may be prone to rupture because of their high lipid content, increased inflammation, and thin fibrous cap. These plaques are categorized morphologically as one of two variants: type IV lesions, consisting of confluent cellular lesions with a great deal of extracellular lipid intermixed with normal intima, which may predominate as an outer layer or cap; or type Va lesions, possessing an extracellular lipid core covered by an acquired fibrous cap. Phase 2 plaques can evolve into the acute phases 3 and 4. Phase 3. These lesions are characterized by acute complicated type VI lesions, originating from ruptured (type IV or Va) or eroded lesions, and leading to mural, non-obstructive thrombosis. This process is clinically silent, but occasionally may lead to the onset of angina . Phase 4. These lesions are characterized by acute complicated type VI lesions, with fixed or repetitive occlusive thrombosis. This process becomes clinically apparent in the form of an acute coronary syndrome (ACS), although not infrequently it is silent . About two-thirds of ACS are caused by occlusive thrombosis on a non-stenotic plaque, although in about one-third, the thrombus occurs on the surface of a stenotic plaque . In phases 3 and 4, changes in the geometry of ruptured plaques, as well as organization of the occlusive or mural thrombus by connective tissue, can lead to the occlusive or significantly stenotic and fibrotic plaques. Phase 5. These lesions are characterized by type Vb (calcific) or Vc (fibrotic) lesions that may cause angina; however, if preceded by stenosis or occlusion with associated ischemia, the myocardium may be protected by collateral circulation and such lesions may then be silent or clinically inapparent.")
Similar presentations
































 Definition of ACS Signs and symptoms of ACS Gender and age related difference in ACS Pathophysiology.>")