Download presentation
Presentation is loading. Please wait.

1
DFT Density Functional theory and the description of Nature
Edison Z. da Silva Instituto de Física "Gleb Wataghin", UNICAMP, Campinas - SP, Brazil

2
Outline DFT, What it is, how it came about, what it is used for.....
Solids and DFT Probing the Earth´s Inner Core Gold Nanowires

3
We have a well defined system
Nuclei + Electrons Molecules Atoms Solids Liquids

4
DFT, theoretical framework
Electrons are the “glue” which holds matter together DFT, theoretical framework Description of known materials, prediction of new materials, explanation of many questions

5
Electromagnetic Forces
Fundamental laws of electronic and nuclear motions well known Electromagnetic Forces + Quantum Mechanics H ( {ri , r} ) = ETOT ( {ri , r} ) ^ H = ( -½2i ) + ( -½ 2 ) + ( - ) + Z ri ___ i<j Ne i=1 ^ 1 rij =1 Nn < ZZ _____ r M Te ^ Tn ^ Ve-n ^ Ve-e ^ Vn-n ^
 = ETOT ( {ri , r} ) ^ H = ( -½2i ) + ( -½ 2 ) + ( - ) + + Z ri ___. i<j. Ne. i=1. ^ 1. rij. =1. Nn. < ZZ _____. r M Te. ^ Tn. ^ Ve-n. ^ Ve-e. ^ Vn-n. ^")
6
“There is an oral tradition that, shortly after Schrödinger’s equation for the electronic wavefunction had been put forward and spectacularly validated for small systems like He and H2, P. A. M. Dirac declared that chemistry had come to an end - its content was entirely contained in that powerful equation. Too bad, he is said to have added, that in almost all cases, this equation was far too complex to allow solution.” W. Kohn, Reviews of Modern Physics 71, 1253 (1999).
.")
7
Therefore, we have a well posed problem, but too complex and difficult to solve
Impossible to have exact solution except in very few cases Need of approximations

8
Born-Oppenheimer (1927) Approximation (Qualitatively)
Based on the fact that nuclear masses are much larger than the electronic ones Therefore, electrons move on a different time-scale than the nuclei (nuclei move much more slowly) Thus, electrons follow the nuclei “instantaneously” This allows us to consider the nuclei fixed when solving for the electronic motion
 Thus, electrons follow the nuclei instantaneously This allows us to consider the nuclei fixed when solving for the electronic motion.")
9
Helecaelec({ri};{r}) = Eaelec({r}) aelec({ri};{r})
Born-Oppenheimer Approximation Helec = ( -½2i ) + ( - ) + Z ri ___ i<j Ne i=1 ^ 1 rij =1 Nn ZZ _____ r < Te ^ Ve-n ^ Ve-e ^ Vn-n ^ Helecaelec({ri};{r}) = Eaelec({r}) aelec({ri};{r}) ^ aelec({ri};{r}) depends explicitly on the electronic coordinates {ri} but only parametrically on the nuclear coordinates {r}, as does Eaelec({r})
 + ( - ) + + Z ri ___. i<j. Ne. i=1. ^ 1. rij. =1. Nn. ZZ _____. r < Te. ^ Ve-n. ^ Ve-e. ^ Vn-n. ^ Helecaelec({ri};{r}) = Eaelec({r}) aelec({ri};{r}) ^ aelec({ri};{r}) depends explicitly on the electronic coordinates {ri} but only parametrically on the nuclear coordinates {r}, as does Eaelec({r})")
10
(r , Q) = a(Q) aelec(r;Q)
Born-Oppenheimer Approximation As the {aelec(r;Q)}, for a fixed Q, form a complete orthonormalized set, one can write: (r , Q) = a(Q) aelec(r;Q) a [Tn + Ebelec(Q)] b(Q) + ba(Q) a(Q) = Etot b(Q) ^ a Roughly speaking, if the kinetic energy of the nuclei is small when compared to |Ebelec(Q) - Eaelec(Q)|, the non-diagonal terms ba can be neglected
}, for a fixed Q, form a complete orthonormalized set, one can write: (r , Q) = a(Q) aelec(r;Q) a. [Tn + Ebelec(Q)] b(Q) + ba(Q) a(Q) = Etot b(Q) ^ a. Roughly speaking, if the kinetic energy of the nuclei is small when compared to |Ebelec(Q) - Eaelec(Q)|, the non-diagonal terms ba can be neglected.")
11
(r , Q) = b(Q) belec(r;Q)
Born-Oppenheimer Approximation [Tn + Ebelec(Q)] b(Q) = Etot b(Q) ^ (r , Q) = b(Q) belec(r;Q) The total electronic energy Ebelec(Q) acts as a potential energy for the nuclear motion, which occurs in a single electronic state belec . The nuclear motion simply deforms the electronic distribution, and does not cause transitions between different electronic states.
] b(Q) = Etot b(Q) ^ (r , Q) = b(Q) belec(r;Q) The total electronic energy Ebelec(Q) acts as a potential energy for the nuclear motion, which occurs in a single electronic state belec . The nuclear motion simply deforms the electronic distribution, and does not cause transitions between different electronic states.")
12
Born-Oppenheimer Approximation
Important concepts: Molecular geometry

13
Born-Oppenheimer Approximation
Crystal structure Important concepts: Molecular geometry

14
Born-Oppenheimer Approximation
Important concepts: Molecular geometry Crystal structure Potential Energy Surface (PES) Geometrical coordinate PES Ex: Carbon Amorphous Graphite Diamond
 Geometrical. coordinate. PES. Ex: Carbon. Amorphous. Graphite. Diamond.")
15
Two issues How to calculate (as accurately as possible) the PES
electronic degrees of freedom How to sample the PES (or how to move the atoms) nuclear degrees of freedom
 nuclear degrees of freedom.")
16
How to solve Helecelec = Eelecelec
Traditional way of thinking would be to describe the wavefunction as well as possible However, WF for a many electron system is a very complicated object!

17
YES Is there another way to solve the problem (ab initio)?
Electronic density e(r) as the fundamental object (and not wavefunction elec)
 as the fundamental object. (and not wavefunction elec)")
18
Density Functional Theory (DFT)
Proc. Cambridge Philos. Soc. 26, 376 (1930) Roots in the works of Thomas (1926), Fermi (1928), Dirac (1930), Slater, etc. Chemistry Nobel prize Formally well founded in the works of W. Kohn P. Hohenberg and W. Kohn, Phys. Rev 136, 864B (1964) W. Kohn and L. J. Sham, Phys. Rev. 140, 1133A (1965)
 Roots in the works of Thomas (1926), Fermi (1928), Dirac (1930), Slater, etc. Chemistry Nobel prize Formally well founded in the works of W. Kohn. P. Hohenberg and W. Kohn, Phys. Rev 136, 864B (1964) W. Kohn and L. J. Sham, Phys. Rev. 140, 1133A (1965)")
19
DFT - Two basic theorems P. Hohenberg and W. Kohn, Phys
DFT - Two basic theorems P. Hohenberg and W. Kohn, Phys. Rev 136, 864B (1964) Let’s consider a system of N interacting electrons in the ground-state associated with an external potential v(r) 1) The ground state density 0e(r) uniquely determines the potential v(r) Since with v(r) the full electronic Hamiltonian is known, 0e(r) completely determines all properties of the system 2) There exists a functional E[e(r)] which has its minimum for the ground-state density 0e(r) Can use this variational principle to calculate 0e(r)
 Let’s consider a system of N interacting electrons in the ground-state associated with an external potential v(r) 1) The ground state density 0e(r) uniquely determines the potential v(r) Since with v(r) the full electronic Hamiltonian is known, 0e(r) completely determines all properties of the system. 2) There exists a functional E[e(r)] which has its minimum for the ground-state density 0e(r) Can use this variational principle to calculate 0e(r)")
20
DFT - How to calculate 0e W. Kohn and L. J. Sham, Phys. Rev
DFT - How to calculate 0e W. Kohn and L. J. Sham, Phys. Rev. 140, 1133A (1965) E[e] = v(r) e(r) d3r + T[e] + U[e] I don’t know how to solve this problem, but I know how to solve the following problem: If I had a system of non-interacting electrons, in an external potential veff(r), then: Es[e] = veff(r) e(r) d3r + Ts[e] Ts[e] = kinetic energy of a non-interacting system with density e
 E[e] = v(r) e(r) d3r + T[e] + U[e] I don’t know how to solve this problem, but I know how to solve the following problem: If I had a system of non-interacting electrons, in an external potential veff(r), then: Es[e] = veff(r) e(r) d3r + Ts[e] Ts[e] = kinetic energy of a non-interacting system with density e.")
21
DFT - How to calculate 0e W. Kohn and L. J. Sham, Phys. Rev
DFT - How to calculate 0e W. Kohn and L. J. Sham, Phys. Rev. 140, 1133A (1965) The ground-state wavefunction in this case is a Slater determinant of N orbitals i (i=1,N), which satisfy the equations: [-½ 2 + veff(r)] i(r) = i i(r) And the ground-state density can be written as: 0e(r) = |i(r)|2 lowest N i
 The ground-state wavefunction in this case is a Slater determinant of N orbitals i (i=1,N), which satisfy the equations: [-½ 2 + veff(r)] i(r) = i i(r) And the ground-state density can be written as: 0e(r) = |i(r)|2. lowest N. i.")
22
DFT - How to calculate 0e W. Kohn and L. J. Sham, Phys. Rev
DFT - How to calculate 0e W. Kohn and L. J. Sham, Phys. Rev. 140, 1133A (1965) However, from the Hohenberg-Kohn theorem, a completely equivalent way of calculating 0e(r) would be through the minimization of E[e(r)]: e(r) Es[e] - e(r’) d3r’ = 0 + veff(r) - = 0 Ts[e] e(r) 0e(r) = |i(r)|2 i Gives same density as
 However, from the Hohenberg-Kohn theorem, a completely equivalent way of calculating 0e(r) would be through the minimization of E[e(r)]: e(r) Es[e] - e(r’) d3r’ = 0. + veff(r) - = 0. Ts[e] e(r) 0e(r) = |i(r)|2. i. Gives same density as.")
23
DFT - How to calculate 0e W. Kohn and L. J. Sham, Phys. Rev
DFT - How to calculate 0e W. Kohn and L. J. Sham, Phys. Rev. 140, 1133A (1965) Application of the Hohenberg-Kohn theorem to E[e] gives e(r) E[e] - e(r’) d3r’ = 0 Ts[e] + v(r’) e(r’) d3r’ + UH[e] + EXC[e] - e(r’) d3r’ e(r) = 0 + v(r) + d3r’e(r’)/|r-r’| + Ts[e] e(r) EXC[e] - = 0
 Application of the Hohenberg-Kohn theorem to E[e] gives. e(r) E[e] - e(r’) d3r’ = 0. Ts[e] + v(r’) e(r’) d3r’ + UH[e] + EXC[e] - e(r’) d3r’ e(r) = 0. + v(r) + d3r’e(r’)/|r-r’| + Ts[e] e(r) EXC[e] - = 0.")
24
DFT - How to calculate 0e W. Kohn and L. J. Sham, Phys. Rev
DFT - How to calculate 0e W. Kohn and L. J. Sham, Phys. Rev. 140, 1133A (1965) Comparison with similar equation for non-interacting electrons gives: + v(r) + d3r’e(r’)/|r-r’| + Ts[e] e(r) EXC[e] - = 0 Interacting electrons + veff(r) - = 0 Ts[e] e(r) Non-interacting electrons v(r) + d3r’e(r’)/|r-r’| + EXC[e] e(r) veff(r) =
 Comparison with similar equation for non-interacting electrons gives: + v(r) + d3r’e(r’)/|r-r’| + Ts[e] e(r) EXC[e] - = 0. Interacting electrons. + veff(r) - = 0. Ts[e] e(r) Non-interacting electrons. v(r) + d3r’e(r’)/|r-r’| + EXC[e] e(r) veff(r) =")
25
DFT - How to calculate 0e W. Kohn and L. J. Sham, Phys. Rev
DFT - How to calculate 0e W. Kohn and L. J. Sham, Phys. Rev. 140, 1133A (1965) We can, therefore, calculate exactly (in principle) the problem of the interacting electrons through the solution of the following equations: [-½ 2 + veff(r)] i(r) = i i(r) 0e(r) = |i(r)|2 lowest N i v(r) + d3r’e(r’)/|r-r’| + EXC[e] e(r) veff(r) =
 We can, therefore, calculate exactly (in principle) the problem of the interacting electrons through the solution of the following equations: [-½ 2 + veff(r)] i(r) = i i(r) 0e(r) = |i(r)|2. lowest N. i. v(r) + d3r’e(r’)/|r-r’| + EXC[e] e(r) veff(r) =")
26
DFT - How to calculate 0e W. Kohn and L. J. Sham, Phys. Rev
DFT - How to calculate 0e W. Kohn and L. J. Sham, Phys. Rev. 140, 1133A (1965) Problem of many-electrons mapped exactly (in principle) into the problem of non-interacting electrons subjected to an effective potential! v(r) + d3r’e(r’)/|r-r’| + EXC[e] e(r) veff(r) = However, we don’t know how to calculate this effective potential: We need approximations for EXC[e] and vXC[e] = EXC[e] e(r)
 Problem of many-electrons mapped exactly (in principle) into the problem of non-interacting electrons subjected to an effective potential! v(r) + d3r’e(r’)/|r-r’| + EXC[e] e(r) veff(r) = However, we don’t know how to calculate this effective potential: We need approximations for EXC[e] and vXC[e] = EXC[e] e(r)")
27
DFT - Local Density Approximation (LDA)
Let’s consider the exchange-correlation energy, per particle, for a homogeneous electron gas, ehXC This quantity can be calculated very accurately (Quantum Monte Carlo) Let’s imagine the real system divided in small cells: Ni Vi i = Ni / Vi i
 Let’s imagine the real system divided in small cells: Ni Vi i = Ni / Vi. i.")
28
DFT - Local Density Approximation (LDA)
If we consider that the density is approximately homogeneous inside cell i, then we can write for the XC energy in cell i EiXC ehXC (i) Ni = ehXC (i) Ni (Vi / Vi) = = ehXC (i) (Ni / Vi) Vi = ehXC (i) i Vi i Ni Vi i = Ni / Vi
 Ni. = ehXC (i) Ni (Vi / Vi) = = ehXC (i) (Ni / Vi) Vi. = ehXC (i) i Vi. i. Ni Vi i = Ni / Vi.")
29
DFT - Local Density Approximation (LDA)
Summing over all the cells i will give for the total XC energy EXC ehXC(i) i Vi i EXC ehXC((r)) (r) d3r = ELDAXC i Ni Vi i = Ni / Vi
 i Vi. i. EXC ehXC((r)) (r) d3r = ELDAXC. i. Ni Vi i = Ni / Vi.")
30
DFT - Local Density Approximation (LDA)
And from this we can get the XC potential vLDAXC ELDAXC e vLDAXC = = ehXC((r)) (r) d3r e = ehXC() d d e i Ni Vi i = Ni / Vi
) (r) d3r. e. = ehXC() d. d e. i. Ni Vi i = Ni / Vi.")
31
DFT - Self-consistency cycle
Still a problem: veff depends one ande depends on veff Solution: self-consistency cycle 1. Choose an initial density 0e and determine the effective potential veff (0e) 2. Solve the Kohn-Sham equations [-½ 2 + veff(r)] i(r) = i i(r) 3. Calculate the density 1e(r) = |i(r)|2 lowest N i 4. Check if 1e = 0e. If yes, end of cycle. If not, go back to 2. (with 0e = 1e)
 2. Solve the Kohn-Sham equations. [-½ 2 + veff(r)] i(r) = i i(r) 3. Calculate the density. 1e(r) = |i(r)|2. lowest N. i. 4. Check if 1e = 0e. If yes, end of cycle. If not, go back to 2. (with 0e = 1e)")
32
Crystal - Bloch functions
veff has the periodicity of the crystal According to Bloch’s theorem, the i will have the following form: (n,k)(r) = exp(i k•r) u(n,k)(r) The function u(n,k) also has the periodicity of the crystal Therefore, it can be expanded in a discrete set of plane waves whose wave vectors are reciprocal lattice vectors of the crystal u(r) = cGexp(i G•r) G
(r) = exp(i k•r) u(n,k)(r) The function u(n,k) also has the periodicity of the crystal. Therefore, it can be expanded in a discrete set of plane waves whose wave vectors are reciprocal lattice vectors of the crystal. u(r) = cGexp(i G•r) G.")
33
Crystal - Bloch functions
Therefore (n,k)(r) = cnk+G exp[i (k+G)•r] G For a crystal, the density will be given by e(r) = |i(r)|2 = 2 |(n,k)(r)|2 lowest N i occ. bands n k BZ
(r) = cnk+G exp[i (k+G)•r] G. For a crystal, the density will be given by. e(r) = |i(r)|2 = 2 |(n,k)(r)|2. lowest N. i. occ. bands. n. k. BZ.")
34
Crystal - Bloch functions
Two approximations: 1) Maximum value of G=GC number of basis function (plane wave) (n,k)(r) = cnk+G exp[i (k+G)•r] G Gc To determine GC one gives the kinetic energy associated with the GC Cutoff energy EC = GC2/2m 2) Number of Brillouin-zone points - set of “special points” e(r) = 2 |(n,k)(r)|2 occ. bands n k SP
 Maximum value of G=GC number of basis function (plane wave) (n,k)(r) = cnk+G exp[i (k+G)•r] G. Gc. To determine GC one gives the kinetic energy associated with the GC. Cutoff energy EC = GC2/2m. 2) Number of Brillouin-zone points - set of special points e(r) = 2 |(n,k)(r)|2. occ. bands. n. k. SP.")
35
Strong oscillations in the core region
Crystal - Bloch functions Strong oscillations in the core region All electron valence WF

36
These strong oscillations are bad for plane-wave expansion (large value of GC to describe small features in real space) Either use a different basis function in the core region, or change the function in the core region

37
Pseudopotentials Valence electrons are the chemically relevant ones
Removes the core electrons and replaces them and the strong potential by an effective, weaker potential that acts on the valence electrons rc r V,

38
All electron Pseudo

39
Si, The credibility of DFT

40
How to solve the KS equations
After all these approximations, I have to find the cnk+G through the self-consistent solution of the KS equations HKS i(r) = i i(r) (n,k)(r) = cnk+G exp[i (k+G)•r] G Gc
 = i i(r) (n,k)(r) = cnk+G exp[i (k+G)•r] G. Gc.")
41
= c HKS Traditional method - diagonalization
Write the matrix for the KS Hamiltonian in the plane wave basis set and diagonalize it HKSk+G,k+G’ cnk+G’ = nk+G cnk+G G’ HKS c =

42
Perfect crystal

43
Crystal with a defect

44
Crystal with a defect

45
Crystal with a defect

46
Surfaces

47
Surfaces

48
Surfaces

49
Applications Understanding the Earth´s inner core
Breaking of Gold nanowires

50
Elasticity of Iron at the temperature of the Earth´s inner core
G. Stainle-Neumann, L. Stixrude, R. Cohen & O. Gulseren Nature, 413, 57 (2001)
")
51
General picture

52
Seismological Studies:
Compressional waves travel faster along polar paths than in equatorial plane. Earth´s center, 2,440 km across is nearly pure Iron At high temperatures and pressures iron goes to an hcp phase called e-phase

53
Iron at Temperatures and pressures of the Earth´s core.
Crystal structure distorts unexpectedly Inner core believed to be made up of Iron crystals in hcp structure Observed seismic anisotropy must arise from differences in elastic response of hcp Iron Densities for iron 7.87 Mg m atmospheric pressure 13.04 Mg m-3 at Earth´s core pressures

54
Iron at Temperatures and pressures of the Earth´s core.
DFT calculations of elastic constants Thermodynamics given by the Helmholtz free energy F(V,T) = E0(V) + Eel(V,T) -T Sel(V,T) + Fvib(V,T) E0(V) static total energy Eel(V,T) due to thermal excitations Sel(V,T) electronic entropy Fvib(V,T) phonon contribution (particle in a cell )
 = E0(V) + Eel(V,T) -T Sel(V,T) + Fvib(V,T) E0(V) static total energy. Eel(V,T) due to thermal excitations. Sel(V,T) electronic entropy. Fvib(V,T) phonon contribution (particle in a cell )")
55
r(Mg m-3) 12.52 13.04 13.62 Equilibrium structure of hcp iron over a range of densities and temperatures of the Earth´s core. Computing F for different c/a and finding the minima

56
Elastic constants Elasticity of hcp iron for a density Mgm-3

57
Velocity Acoustic properties of iron for the Earth´s core.
Adiabatic bulk modulus (KS) and shear modulus (m) as function of temperature compared with inner core at the same density Mgm-3 Travel time anomaly Solid line , model with 1/3 of crystals with basal planes aligned with rotation axis
 and shear modulus (m) as function of temperature. compared with inner core at. the same density Mgm-3. Travel time anomaly. Solid line , model with 1/3. of crystals with basal planes. aligned with rotation axis.")
58
Conclusions Seismological Studies:
Compressional waves travel faster along polar paths than in equatorial plane. At high temperatures and pressures iron goes to an hcp phase called e -phase. With c/a = 1.7 seismic waves travel 12 % faster in the ab plane. This anisotropy is too strong, if crystals are aligned it accounts for the experimental findings.

59
Breaking of Gold Nanowires: A Computer Simulation study
E. Z. da Silva Instituto de Física "Gleb Wataghin", UNICAMP, Campinas - SP, Brazil

60
Participants, motivation...
Theory Antonio José R. da Silva(IF-USP) Adalberto Fazzio (IF-USP) Frederico D. Novaes (PhD-IF-USP) Experimental motivation Daniel Ugarte (LNLS) Varlei Rodrigues
 Adalberto Fazzio (IF-USP) Frederico D. Novaes (PhD-IF-USP) Experimental motivation. Daniel Ugarte (LNLS) Varlei Rodrigues.")
61
When we get to the very, very small
world - say circuits of seven atoms - we have a lot of new things that would happen that represent completelly new opportunities for design. Atoms on a small scale behave like nothing on the large scale, for they satisfy the laws of quantum mechanics. Richard Feynman Plenty of Room at the Bottom(1959)
")
62
Program Why study Au nanowires? Experiments Theory TBMD simulations
Impurities Conclusions

63
Why study Au nanowires? Gold nanowires are produced in the lab!
Understanding of its properties is fundamental. Gold is the electrical contact in nanotechnology. When Gold nanowires become very thin, interesting surprises are found: Conductivity is quantized. Possibility of helical structures. One-atom thick wires are produced.

64
Gold in devices J Reichert, et al., Phys. Rev. Lett., 88, (2002)
")
65
Experiments, How it all started!
Pin-plate experiment: nanowire formed conductance during elongation and contraction U.Landman, W.D.Luedtke, B.E.Salisbury, and R.L.Whetten, Phys. Rev. Lett. 77, 1362, (1996)
")
66
Theoretical Predictions
Computer simulations with a “glue” type empirical many-body potential (for Al and Pb) predicted: As the diameter decrease, thin metal wires develop exotic stable noncrystalline structures: Existence of critical diameter Formation of helical, spiral-structured wires O. Gulseren, F Ercolessi, and E. Tosatti, Phys. Rev. Lett. 80, 3775, (1998)
 predicted: As the diameter decrease, thin metal wires develop. exotic stable noncrystalline structures: Existence of critical diameter. Formation of helical, spiral-structured wires. O. Gulseren, F Ercolessi, and E. Tosatti, Phys. Rev. Lett. 80, 3775, (1998)")
67
Theory Weird Nanowires
O. Gulseren, F Ercolessi, and E. Tosatti, Phys. Rev. Lett. 80, 3775, (1998)
")
68
How to make a wire

69
Theorist´s view: production and imaging process!

70
Y. Kondo and K. Takayanagi, Phys. Rev. Lett. 79, 3455, (1997)
Stable Au Nanowires from surface structures Production Au thin films (3nm) in UHVTEM electron beam irradiation (100 A/cm2) produced many holes nanowires Imaging Straight nanowires images produced with electron beam irradiation (5 A/cm2) Y. Kondo and K. Takayanagi, Phys. Rev. Lett. 79, 3455, (1997)
 in UHVTEM. electron beam irradiation (100 A/cm2) produced many holes nanowires. Imaging. Straight nanowires. images produced with electron. beam irradiation (5 A/cm2) Y. Kondo and K. Takayanagi, Phys. Rev. Lett. 79, 3455, (1997)")
71
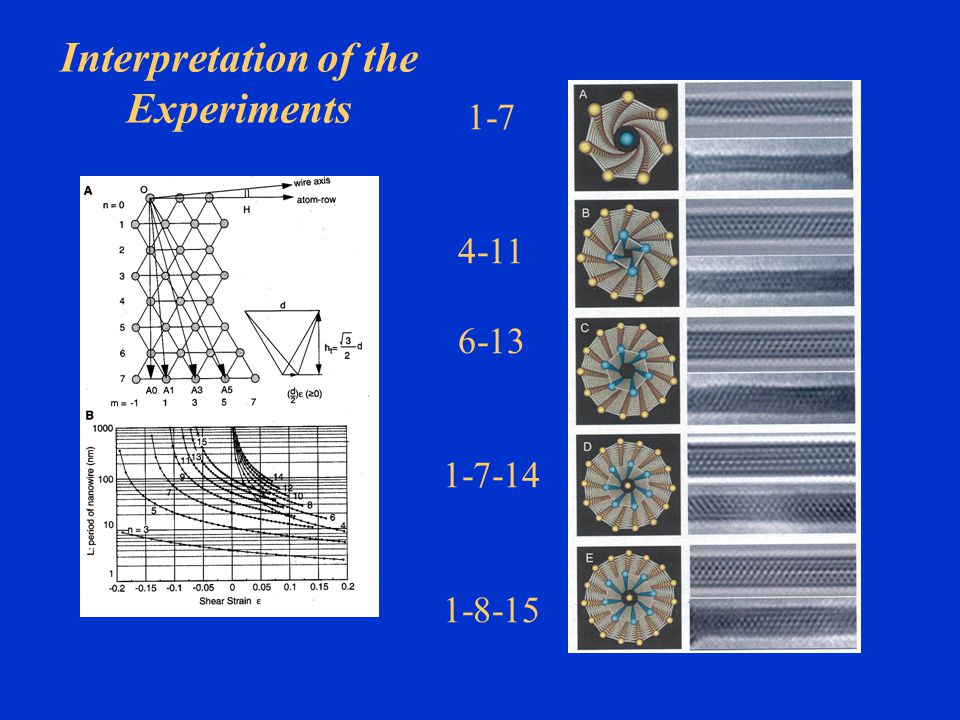
Interpretation of the Experiments
1-7 4-11 6-13 1-7-14 1-8-15
72
Folding of a (111) plane Cylindrical folding of a
triangular lattice for an (m, n) tube, with views of several coaxial tube nanowires. Each atom is pictured as a sphere of atomic radius. The (7, 3) gold nanowire (note its chirality) was reported to be magic in (3).
 tube, with. views of several coaxial. tube nanowires. Each atom. is pictured as a sphere of. atomic radius. The (7, 3) gold. nanowire (note its chirality) was reported to be magic in (3).")
73
V. Rodrigues and D. Ugarte, Phys. Rev. B 63, 073405-1, (2001)
Real time imaging of one-atom-thick Au nanowires The film irradiation technique from Kondo and Takayanagi produced stable one-atom-thick Au- nanowires. Time sequence of formation, elongation and fracture of a chain of gold atoms. V. Rodrigues and D. Ugarte, Phys. Rev. B 63, , (2001)
")
74
Dynamical Simulations of Gold nanowires
Is it possible to simulate the evolution of a gold nanowire under stress with a reliable electronic structure method? Wire under tension. It forms, starts to thin ( atoms going to the wire edges) neck is produced (one atom constriction). Atoms from the wire,( near the tips) are draw back and are incorporated into the one -atom thick neck. This thin neck grows to 4-5 atoms The wire finally breaks
 neck is produced (one atom constriction). Atoms from the wire,( near the tips) are draw back and. are incorporated into the one -atom thick neck. This thin neck grows to 4-5 atoms. The wire finally breaks.")
75
(three orders of magnitude faster)
Ab Initio DFT Methods (costly in terms of computer time!) NRL-TBMD keeps the electrons (three orders of magnitude faster) Number of atoms ACCURACY MD with Classical Potentials Embedded-atom, Effective medium, Finnis-Sinclair, Second Moment(TB) (No electrons!)
 NRL-TBMD. keeps the electrons. (three orders of magnitude faster) Number of atoms. ACCURACY. MD with Classical Potentials. Embedded-atom, Effective medium, Finnis-Sinclair, Second Moment(TB) (No electrons!)")
76
The Tight -Binding Method
Slater and Koster proposed in 1954 a modified combination of atomic orbitals (LCAO) as a interpolation scheme to determine energy bands. In 1986 Papaconstantopoulos published a handbook of SK parameters for most elements in the periodic table. As DFT ab initio methods improve, so does the Papaconstantopoulos TB fits with updates the of SK parameters for most elements in the periodic table.
 as a. interpolation scheme to determine energy bands. In 1986 Papaconstantopoulos published a handbook. of SK parameters for most elements in the periodic table. As DFT ab initio methods improve, so does the. Papaconstantopoulos TB fits with updates the. of SK parameters for most elements in the periodic table.")
77
The NRL tight-binding method
Procedure, The method fits TB parameters to reproduce ab initio total energies (DFT) calculated with full-potential linear aumented plane wave method. Slater-Koster parameters are fitted to LAPW bands and total energies as function of volume for 10 fcc structures, 6 bcc structures and 5 sc structures. Perdue-Wang parametrization to LDA M.J.Mehl and D. A.Papacontantopoulos, Phys. Rev. B 54, 4519 (1996)
 calculated with full-potential linear aumented plane wave method. Slater-Koster parameters are fitted to LAPW bands and. total energies as function of volume for 10 fcc structures, 6 bcc structures and 5 sc structures. Perdue-Wang parametrization to LDA. M.J.Mehl and D. A.Papacontantopoulos, Phys. Rev. B 54, 4519 (1996)")
78
Tight-binding and Molecular Dynamics
TB basis set The Slater-Koster (SK) parameters Tight-binding Fit of the ab initio Potential energy surface Ab initio molecular dynamics
 parameters. Tight-binding Fit of the ab initio Potential energy surface. Ab initio molecular dynamics.")
79
TB basis set Minimum basis set of atomic like orbitals,
for transition metals; Number of basis functions per atom N0. Number of atoms Na, dimension space spanned by this basis set Nb = N0 Na. States are expressed as linear combinations of these orbitals:

80
The Slater-Koster parameters
Matrix elements of the Hamiltonian and overlap gives: Hopping terms Overlap terms SK-parameters

81
Tight-Binding Fit of the ab initio Potential energy surface
Formalism Parametrization

82
Complicated Transition Metal-Mn
Manganese crystallizes in the -Mn structure with 29 atoms Above 710 0C, Mn transforms to the -Mn with 20 atoms Calculated TB total energy

83
Fitting Procedure for Au, using the static TB code
Equation of state How good is the TB parametrization for fcc bulk Au? Data basis: 10 fcc structures, 6 bcc structures, 5 sc structures. Phonon dispersion, T = 0 Fcc structure, lowest energy F.Kirchhhoff et al. Phys. Rev. B, (2001)
")
84
Ab initio molecular dynamics steps of the simulation
Input, Ri, positions of the atoms H the Hamiltonian The electronic ground state of the system is calculated. Eigenvectors Forces on the atoms. Forces (EOM) integrated and new positions found. Where states are expanded using the TB orbitals
 integrated. and new positions found. Where states are expanded using the TB orbitals.")
85
Electronic DOS for Au, using TBMD code
Solid, T dependence MD simulations: Microcanonical ensemble, EM with Verlet algo, t = 2 fs, point sampling. Liquid Au, T = 1773 K F.Kirchhhoff et al. Phys. Rev. B, (2001)
")
86
Au-nanowires, simulations E. Z. da Silva. A J. R. da Silva and A
Au-nanowires, simulations E. Z. da Silva. A J. R.da Silva and A. Fazzio, Phys. Rev. Lett., 87 , (2001) Front view Starting structures: nanowire along the along the(111) direction - 10 atom planes - 7atoms in each plane. Top view
 Front view. Starting structures: nanowire along the. along the(111) direction atom planes. - 7atoms in each plane. Top. view ")
87
Simulation Protocol EM integrated using Verlet algo, t =1 fs
The system is heated to T = 600 K, MD steps, 7ps Elongation of the wire by L = 0.5 Å The system is heated to T = 400 K, 4000 MD steps, 4ps Previous two steps are repeated Procedure done for Å < LW < 44.0 Å

88
Au nanowire, 6 (111) planes of 7 Au atoms,
unrelaxed structure relaxed structure

89
Similarities for the breaking the one-atom necklace
Experiment 2.99 2.98 2.89 2.74 2.63 2.91 2.61 2.89 Simulation

90
Evolution and breaking
Atomic configurations (in Å) (a) 25.5, (b) 33.0, (c) 37.0 (a) 38.0, (b) 40.5, (d) 41.0
 (a) 25.5, (b) 33.0, (c) (a) 38.0, (b) 40.5, (d)")
91
Defect structure, one atom neck
(i) Defect structure (iii) 4-5 atoms planes (iv) atoms planes (v) one atom neck
 Defect structure. (iii) 4-5 atoms planes. (iv) atoms planes. (v) one atom neck.")
92
Forces Saw-tooth behavior

93
Before breaking the one-atom necklace
The nanowire develops a one-atom thick necklace with five atoms in its final structure. Just before breaking, the apex-apex distance gets to da-a ~ Å.

94
Animation of the simulation
See this animation at

95
Pending problems Are TBMD results reproducible using ab initio DFT.
Large Au-Au distances seen in the experiments

96
Real time imaging of one-atom-thick Au nanowires
HRTEM image of atom chain with three gold atoms. Atomic positions are the dark spots Large Au-Au distances!!!! Schematic representation V. Rodrigues and D. Ugarte, Phys. Rev. B 63, , (2001)
")
97
Simulations give shorter Au-Au distances
Before breaking: one-atom necklace Simulations give shorter Au-Au distances

98
Ab initio DFT (Siesta) Fully self-consistent DFT LCAO
Extremely fast simulation using minimal basis set Kohn-Sham equations Exchange-correlation (GGA) Norm conserving pseudopotentials (Troullier-Martins) Total energy Forces
 Norm conserving pseudopotentials (Troullier-Martins) Total energy. Forces.")
99
Ab initio evolution Starting structure Before breaking
Au-Au distance 3.0 Å After breaking

100
Large distances, Impurities?
The statistics The Images Au-Au distances 3.6±0.2 (Au-Au)Max 3.1 Å (Au-X-Au)Max 3.6 Å X = ? Legoas,Galvão, Rodrigues , Ugarte , PRL, 88, , (2002)
Max 3.1 Å. (Au-X-Au)Max 3.6 Å. X = Legoas,Galvão, Rodrigues , Ugarte , PRL, 88, , (2002)")
101
Reasoning for impurities
Structure simulation TEM simulation 2.9 2.8 3.6 3.1

102
Carbon, structure right before breaking
Carbon in Au nanowire Carbon, initial structure Au-nanowire Carbon intermediate structure, 3.73 Carbon, structure right before breaking Quebra

103
Considered the effect of the impurities, H, B, C, N, O and S
Impurities and the Au-Au Bond Length Frederico D. Novaes, A.J.R. da Silva, E.Z. da Silva and A.Fazzio, Phys. Rev. Lett. 90, (2003)22 january Use ab initio DFT to study contamination of the pure Au nanowire by impurities. Considered the effect of the impurities, H, B, C, N, O and S Experiment gives Au-?-Au = 3.6 Å Au-??-Au = 4.8 Å
22 january. Use ab initio DFT to study contamination of the pure Au nanowire by impurities. Considered the effect of the impurities, H, B, C, N, O and S. Experiment gives Au- -Au = 3.6 Å. Au- -Au = 4.8 Å.")
104
Contamination by H and S

105
H Impurities? 3.5 3.5 4.9 Our simulation Experiments:
Legoas,Galvão, Rodrigues , Ugarte , PRL, 88, , (2002) 4.9 3.5
")
106
General Conclusions Future: Importance of Gold nanowires.
Simulations can help understand the dynamical evolution and finally the breaking of metal nanowires. TBMD is reliable and fast. Can be used to study dynamical evolution of metal nanowires. Future: Study of other directions for wire formation. Investigation of nanowires of other metals (Ag, Pt etc..) Use nanowires and their tips to design devices.
 Use nanowires and their tips to design devices.")
107
Conclusions DFT, A effective way of studying and projecting interesting materials. Solids and DFT Probing the Earth´s Inner Core Gold Nanowires

Similar presentations







 describe solids, liquids and gases in terms of the spacing, ordering and motion of atoms or molecules; (b) describe.>")
 states the three states of matter.>")










