Download presentation
Presentation is loading. Please wait.

1
Medical Pharmacology Course Anti-Neoplastic Chemotherapy Issam Makhoul M.D. Hematology/Oncology Division Internal Medicine Department University of Arkansas for Medical Sciences October 3, 2007

2
I-Biology of cancer

3
When Strength is Weakness Integrity and diversity of tissues can be traced to the autonomy and versatility of cells The same autonomy and versatility can lead to cancer by –Allowing access to information that is usually denied to the cells –Corruption of the genetic information –This would divert the cells to acquire novel highly unusual phenotypes that may be incompatible with the normally assigned roles of these cells in organismic structure and physiology Cancers are tissue growths that arise in the wrong space and time

4
Risk of cancer is low at the cellular level At steady state, an adult human body has more than 10 13 cells The number of cells in average human lifetime is 10 16 The risk of having cancer is around 10% Knowing that cancer is a clonal process, the risk of developing cancer at the cellular level is 1/10 17 Cancer is an Inefficient Process

5
1. Adapted from Hanahan D, et al. Cell. 2000;100:57-70. Evading apoptosis Self-sufficiency in growth signals Tissue invasion and metastasis Limitless replicative potential Sustained angiogenesis Insensitivity to antigrowth signals Cancer cells Hallmarks of Cancer

6
Benign vs. Malignant Tumors Benign tumors grow locally without invading neighboring tissues or spreading metastases –Most tumors are benign –Harmless –Occasionally, might be symptomatic due to their hormonal secretions spatial Malignant tumors invade locally and spawn metastases –Invasion of the basement membrane –Spreading metastases through the lymphatic and vascular systems

7
Hallmarks of Cancer Self sufficiency in growth signals Insensitivity to antigrowth signals Limitless replicative potential Evasion of programmed cell death (apoptosis) Sustained angiogenesis Tissue invasion and metastasis Evasion of elimination by the immune system Acquisition of genomic instability
 Sustained angiogenesis Tissue invasion and metastasis Evasion of elimination by the immune system Acquisition of genomic instability")
8
Potential Consequences of EGFR Dysregulation Signaling cascades EGFR PI3K MAPK Nucleus Gene activation Cell cycle progression M G1G1 S G2G2 Myc Fos Jun P P MAPK = mitogen-activated protein kinase. Roskoski. Biochem Biophys Res Commun. 2004;319:1. Rowinsky. Annu Rev Med. 2004;55:433. Survival Proliferation Angiogenesis Invasion Apoptosis Metastasis

9
Hallmarks of Cancer Self sufficiency in growth signals Insensitivity to antigrowth signals Limitless replicative potential Evasion of programmed cell death (apoptosis) Sustained angiogenesis Tissue invasion and metastasis Evasion of elimination by the immune system Acquisition of genomic instability
 Sustained angiogenesis Tissue invasion and metastasis Evasion of elimination by the immune system Acquisition of genomic instability")
10
Insensitivity to Antigrowth Signals Two types: –Soluble growth inhibitors (TGF- /TGF- R) –Immobilized inhibitors on extra-cellular matrix and/or surface of nearby cells Antigrowth signals function by one of tow mechanisms: –By forcing the cells out of the active proliferative cycle into G0 –By inducing differentiation Role of c-myc –Myc-Mad differentiation –Myc-Max proliferation
 –Immobilized inhibitors on extra-cellular matrix and/or surface of nearby cells Antigrowth signals function by one of tow mechanisms: –By forcing the cells out of the active proliferative cycle into G0 –By inducing differentiation Role of c-myc –Myc-Mad differentiation –Myc-Max proliferation")
11
Hallmarks of Cancer Self sufficiency in growth signals Insensitivity to antigrowth signals Limitless replicative potential Evasion of programmed cell death (apoptosis) Sustained angiogenesis Tissue invasion and metastasis Evasion of elimination by the immune system Acquisition of genomic instability
 Sustained angiogenesis Tissue invasion and metastasis Evasion of elimination by the immune system Acquisition of genomic instability")
12
Figure 10.13b The Biology of Cancer (© Garland Science 2007) Telomeres and DNA Senescence Crisis
 Telomeres and DNA Senescence Crisis")
13
Consequences Of Telomere Shortening

14
Figure 10.14a The Biology of Cancer (© Garland Science 2007)
")
15
Telomerases are Upregulated in Cancers Telomerases are enzymes that counteract incomplete replication at the end of chromosomes, by lengthening telomeres Persistent or expression of telomerase activity immortalizes cells

16
Hallmarks of Cancer Self sufficiency in growth signals Insensitivity to antigrowth signals Limitless replicative potential Evasion of programmed cell death (apoptosis) Sustained angiogenesis Tissue invasion and metastasis Evasion of elimination by the immune system Acquisition of genomic instability
 Sustained angiogenesis Tissue invasion and metastasis Evasion of elimination by the immune system Acquisition of genomic instability")
17
Evasion of Programmed Cell Death (Apoptosis) Resistance to apoptosis is a common trait to cancers –Mutation of p53 –PI3 kinase-AKT/PKB hyperactivation –Upregulation of decoy receptor for FAS-L –Overexpression of BCL2
 Resistance to apoptosis is a common trait to cancers –Mutation of p53 –PI3 kinase-AKT/PKB hyperactivation –Upregulation of decoy receptor for FAS-L –Overexpression of BCL2")
18
Hallmarks of Cancer Self sufficiency in growth signals Insensitivity to antigrowth signals Limitless replicative potential Evasion of programmed cell death (apoptosis) Sustained angiogenesis Tissue invasion and metastasis Evasion of elimination by the immune system Acquisition of genomic instability
 Sustained angiogenesis Tissue invasion and metastasis Evasion of elimination by the immune system Acquisition of genomic instability")
19
The Angiogenic Switch 1-2 mm Angiogenic Switch Small tumor Nonvascular Dormant Larger tumor Vascular Metastatic potential

20
Hallmarks of Cancer Self sufficiency in growth signals Insensitivity to antigrowth signals Limitless replicative potential Evasion of programmed cell death (apoptosis) Sustained angiogenesis Tissue invasion and metastasis Evasion of elimination by the immune system Acquisition of genomic instability
 Sustained angiogenesis Tissue invasion and metastasis Evasion of elimination by the immune system Acquisition of genomic instability")
21
Table 14.2 The Biology of Cancer (© Garland Science 2007) Activation of Epithelial- Mesenchymal Transition (EMT)
 Activation of Epithelial- Mesenchymal Transition (EMT)")
22
Table 14.3 The Biology of Cancer (© Garland Science 2007) Transcription Factors Orchestrating the EMT
 Transcription Factors Orchestrating the EMT")
23
Cancers Arise From Stem Cells Stem cells possess: –Self-renewal capacity (immortality) –Differentiation capacity (organ repair and replacement of lost cells) Cancer is a defect of stem cell control –Defect of patterning (Defect of organ formation) –It involves both the stem cells and their stroma
 –Differentiation capacity (organ repair and replacement of lost cells) Cancer is a defect of stem cell control –Defect of patterning (Defect of organ formation) –It involves both the stem cells and their stroma")
24
II-The Cell Cycle

25
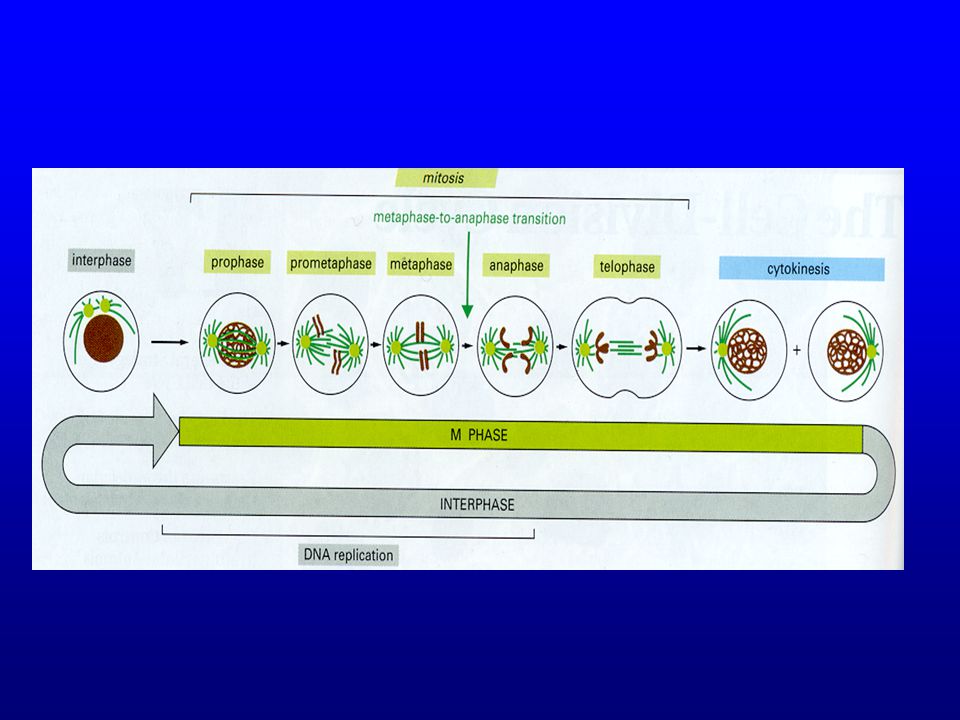
The Cell Cycle It is the ability of a cell to produce exact replicas of itself Cell cycle phases are: –Resting stage (G0) –Interphase G1=active gene expression S=DNA synthesis, DNA doubles 1N 2N G2=post synthetic phase –Mitosis phase or M-phase
 –Interphase G1=active gene expression S=DNA synthesis, DNA doubles 1N 2N G2=post synthetic phase –Mitosis phase or M-phase")
29
III-Chemotherapy for cancer Genetic make up of cancers differs from their normal counterparts only by less than 0.5% The difference in gene expression is often quantitative rather than qualitative Chemotherapy cannot discriminate between normal and cancer cells (risk of toxicity) Cancer cells need longer time than normal cells to recover from injury induced by chemotherapy
 Cancer cells need longer time than normal cells to recover from injury induced by chemotherapy")
30
Choice of chemotherapy The choice of chemotherapy depends on factors related to –A. the tumor –B. the host –C. the drugs

31
A. Tumor Organ of origin and type of cell Cancers are clonal processes Clonal evolution Growth fraction Growth rate

32
Tumors Keep Features of the Differentiated Tissue From Which They Arose Epithelia: carcinomas –Squamous cell carcinomas –Adenocarcinomas Mesenchymal cells –Sarcomas Hematopoietic cells –Leukemias and lymphomas Central and peripheral NS –Neuroectodermal tumors Non classified –SCLC

33
Figure 2.17 The Biology of Cancer (© Garland Science 2007)
")
34
Exponential cell kinetic (Skipper model) L1210 murine leukemia Growth rate is constant and growth fraction > 90% Doubling time forms a straight line on a semilog plot The cytotoxic effects of cancer drugs in this tumor model follow log cell-kill kinetics The closest human tumors to this model are trophoblastic neoplasms, germ cell tumors and Burkitts lymphoma
 L1210 murine leukemia Growth rate is constant and growth fraction > 90% Doubling time forms a straight line on a semilog plot The cytotoxic effects of cancer drugs in this tumor model follow log cell-kill kinetics The closest human tumors to this model are trophoblastic neoplasms, germ cell tumors and Burkitts lymphoma")
35
The concept of log-cell kill is the rationale for giving multiple cycles of chemotherapy to eliminate the tumor and achieve cure.

36
The Gompertzian Model The growth fraction of the tumor is not constant Growth rate peaks when the tumor is approximately 37% of its maximum size Response to chemotherapy in drug-sensitive tumors depends on where the tumor is on its particular growth curve Sensitive Gompertzian-growing tumors respond to cytotoxic drugs in a Gompertzian fashion

38
Explanation of the Gompertzian model Skipper model applies only to proliferating cells Expanding solid tumors outgrow their blood supply leading to anoxia and slowing of the cell cycle Certain cells exit the cell cycle into G0 or resting phase Resting cells become temporarily resistant to chemotherapy (kinetic resistance to chemotherapy)
")
39
How can we increase cell cycling in a given tumor? Debulking surgery

40
Resistance to chemotherapy Two types: kinetic resistance and genetic resistance Kinetic resistance, by entering G0: –Temporary –Reversible when the cell reenters the cell cycle Genetic resistance, by mutation: –Malignant cells are genetically unstable –Malignant cells have higher rate of mutation than normal cells (10 -5 vs. 10 -6 ) –Resistance to chemotherapy occurs in mutant clones: The # of drug-resistant clones // the length of time the tumor has been present or the # of cell divisions
 –Resistance to chemotherapy occurs in mutant clones: The # of drug-resistant clones // the length of time the tumor has been present or the # of cell divisions.")
41
The Goldie-Coldman hypothesis Resistance to two drugs should be less likely than 1/10 10 Multi-drug chemotherapy should be more effective than single-drug therapy

42
Combination chemotherapy Drugs used in combination therapy should –Be active as single agents –Have different mechanisms of action –Have different dose-limiting toxicity –Have different patterns of resistance –Be used at their optimal doses and schedule, with toxicity being the dose limiting factor

43
Mechanisms of resistance to chemotherapy Decreased drug transport into cells (methotrexate…) Increased drug transport out of cells by increased expression of MDR gene (vinca alkaloids, etoposide and anthracyclines) Reduced activation of drug (cytarabine, 6-MP, 6-TG, methotrexate…) Increased inactivation of drug or active metabolite (cytarabine…) Increased DNA repair (all alkylating agents, antitumor antibiotics, topo.II inhibitors)
 Increased drug transport out of cells by increased expression of MDR gene (vinca alkaloids, etoposide and anthracyclines) Reduced activation of drug (cytarabine, 6-MP, 6-TG, methotrexate…) Increased inactivation of drug or active metabolite (cytarabine…) Increased DNA repair (all alkylating agents, antitumor antibiotics, topo.II inhibitors)")
44
Mechanisms of resistance to chemotherapy Use of alternate pathways as source of metablolites (methotrexate, 5-FU, 6-MP, 6-TG…) Gene amplification of enzyme target of drug action (methotrexate, 5-FU, 2-deoxycoformycin) Loss of apoptosis (loss of p53 or bcl2 expression in a tumor cancer cell can lead to resistance to all chemotherapeutic agents). Alteration of target to reduce drug binding (vinvristine, methotraxate, 5-FU, hydeoxyurea, doxo)
.")
45
B. The host Non-selectivity of cancer drugs results in toxicity to normal tissues especially rapidly dividing cells (bone marrow, skin, GI tract and hair follicles) Supportive care –Recombinant growth factors (G-CSF, GM-CSF, erythropoietin) –Blood product transfusion (RBC, platelet) –The use of modern antibiotics Normal tissues never develop resistance The effect of chemotherapy on normal tissue is cumulative Most chemotherapy agents are metabolized in the liver and excreted by the kidney
 Supportive care –Recombinant growth factors (G-CSF, GM-CSF, erythropoietin) –Blood product transfusion (RBC, platelet) –The use of modern antibiotics Normal tissues never develop resistance The effect of chemotherapy on normal tissue is cumulative Most chemotherapy agents are metabolized in the liver and excreted by the kidney.")
46
C. Drugs Chemotherapy agents differ in their –mechanism of action –Spectrum –dose and route of administration Pharmacokinetics is –the study of the drug absorption, distribution, metabolism, and excretion The area under the curve (AUC) in a plot of in vivo concentration versus time IS –The most important parameter in dose level
 in a plot of in vivo concentration versus time IS –The most important parameter in dose level.")
47
C. Drugs Pharmacodynamics –the study of dose-effect relationships or the action of the drug on the body and includes both toxic and therapeutic effects Chemotherapy agents show a steep dose-response curve and modest dose reduction may lead to substantial reductions in tumor cell kill Chemotherapy agents have narrow therapeutic window Increase of dose intensity by: –Regional administration of a drug –Bone marrow rescue

48
IV- Antineoplastic agents

49
Mechanism of action Agents that damage the DNA template by: –Alkylation Nitrogen mustards Nitrosoureas Others –Platinum crosslinking –DNA double-strand cleavage via topoisomerase II: Antibiotics Podophyllotoxins: etoposide, teniposide –DNA single-strand cleavage via topoisomerase I: topotecan, irinotecan –Intercalation, blocking DNA synthesis: dactinomycin, mithramycin

50
Mechanism of action Spindle poisons: –Vinca alkaloids: vincristine, vinblastine, vendesine, vinorelbine –Taxanes: taxol, taxotere

51
Mechanism of action Antimetabolites (enzyme inhibitors) –Thymidylate synthase: 5-fluorouracil –Dihydrofolate reductase: methotrexate –DNA polymerase: cytosine arabinoside (cytarabine), fludarabine –Ribonucleotide reductase: hydoxyurea –Phosphoribosylpyrophosphate amidotransferase: 6-MP, 6-TG –Adenosine deaminase: deoxycoformycin
 –Thymidylate synthase: 5-fluorouracil –Dihydrofolate reductase: methotrexate –DNA polymerase: cytosine arabinoside (cytarabine), fludarabine –Ribonucleotide reductase: hydoxyurea –Phosphoribosylpyrophosphate amidotransferase: 6-MP, 6-TG –Adenosine deaminase: deoxycoformycin")
52
Mechanism of action Hormonal and antihormonal agents: –Agonists: estrogens, androgens, progestins, corticosteroids –Antagonists: tamoxifen, amioglutethimide, leuprolide, flutamide –Aromatase inhibitors: anastrozole, letrozol, examestene

53
Mechanism of action Biologic response modifiers: –Interferons, interleukins, BCG Differentiation agents –All trans-retinoic acid (ATRA) Miscellanous: –Mitotane, asparaginase, prednimustine, estramastine
 Miscellanous: –Mitotane, asparaginase, prednimustine, estramastine")
54
Cell Cycle Specificity

55
Alkylating agents: The most important classes of alkylating agents are –Nitrogen mustards (mechlorethamine, cyclophosphamide, ifosfamide, melphalan, chlorambucil) –Alkyl sulfonates (busulfan) –Nitosoureas (BCNU, CCNU, MeCCNU)
 –Alkyl sulfonates (busulfan) –Nitosoureas (BCNU, CCNU, MeCCNU)")
56
Properties Add functional groups (alkyl groups) to nitrogen (an SN2 reaction) The 7 nitrogen atom of guanine is most susceptible DNA is the most common molecule alkylated These agents will also alkylate proteins Exert the most effect on proliferating cells Alkylating agents are not cell cycle phase specific
 to nitrogen (an SN2 reaction) The 7 nitrogen atom of guanine is most susceptible DNA is the most common molecule alkylated These agents will also alkylate proteins Exert the most effect on proliferating cells Alkylating agents are not cell cycle phase specific")
57
Properties Chemical characteristics of alkylating agents –Mechlorethamine: the least stable, rapidly metabolized –Cyclophosphamide: inactive in its parent compound and needs to be activated by hepatic cytochrome P450 –Melphalan: stable –Chlorambucil: stable

58
Consequences of alkylation Alkylation of guanine causes –Mispairing: instead of G+C, G will pair with T and point mutation will be introduced during DNA replication (G+C T+A) –Cross-linking bifunctional alkylating agents can Cross-link DNA and interfere with replication. Cause covalent linkage between proteins and DNA and thus interfere with transcription

59
Clinical characteristics of alkylating agents Clinical uses: –Hodgkins disease, lymphomas, multiple myeloma, carcinoma of the breast, ovary, melanoma, peripheral stem cell mobilization Toxicities: –myelosuppression –hemorrhagic cystitis (cyclophophamide, probably acrolein), –sterility –secondary leukemia (5-7 years latency, mutations of 5, 7 or 8 chromosomes)
, –sterility –secondary leukemia (5-7 years latency, mutations of 5, 7 or 8 chromosomes)")
60
Antibiotics Anthracyclines (Isolated from Streptomyces peuceitus) –Daunorubicin –Doxorubicin –Idarubicin –Mitoxantrone Bleomycin (Isolated from Streptomyces veicillus) Dactinomycin (Isolated from Streptomyces species) Mitomycin C (Isolated from Streptomyces caespitosus)
 –Daunorubicin –Doxorubicin –Idarubicin –Mitoxantrone Bleomycin (Isolated from Streptomyces veicillus) Dactinomycin (Isolated from Streptomyces species) Mitomycin C (Isolated from Streptomyces caespitosus)")
61
Mechanism of action of antibiotics Anthracyclines (daunorubicin, doxorubicin, idarubicin, mitoxantrone) –intercalate into DNA and prevent replication –can react with cytochrome P450 to generate free radicals, which requires the presence of iron element Mitoxantrone (synthetic anthracenedione) generates fewer free radicals lower toxicity to normal tissues, especially cardiac cells. These molecules are cytotoxic by inducing DNA strand breaks (topoisomerase II inhibitors)
.")
62
Mechanism of action of antibiotics Bleomycin: –It binds to iron (Fe ++ ) and react with molecular oxygen to cause free radical formation which induces DNA strand breaks Dactinomycin: –It intercalates between guanosine-cytosine pairs in DNA, blocking transcription by DNA-dependent RNA polymerase Mitomycin C: –It alkylates the DNA by cross-linking DNA at the N 6 position of adenine and the N 7 position of guanine
 and react with molecular oxygen to cause free radical formation which induces DNA strand breaks Dactinomycin: –It intercalates between guanosine-cytosine pairs in DNA, blocking transcription by DNA-dependent RNA polymerase Mitomycin C: –It alkylates the DNA by cross-linking DNA at the N 6 position of adenine and the N 7 position of guanine")
63
Clinical characteristics of antibiotics: Anthracyclines: –Clinical use: acute leukemia, lymphomas, breast cancer, sarcomas –Toxicities: cardiotoxicity (cardiomyopathy) myelosuppression, vesicants Bleomycin: –Clinical use: testicular cancer, lymphomas, Hodgkins disease –Toxicities: pulmonary fibrosis
 myelosuppression, vesicants Bleomycin: –Clinical use: testicular cancer, lymphomas, Hodgkins disease –Toxicities: pulmonary fibrosis")
64
Clinical characteristics of antibiotics: Dactinomycine: –Clinical use: Wilms tumors, rhabdomyosarcoma, Kaposis sarcoma, gestational choriocarcinoma, Hodgkins disease –Toxicity: myelosuppression Mitomycin C –Clinical use: GI, head and neck, anal cancer –Toxicities: late myelosuppression

65
Antimetabolites Folic acid analogs Pyrimidine analogs Purine analogs

66
Mechanism of action These compounds substitute an enzyme substrates or cofactors and inhibit purine or pyrimidine biosynthesis. They are S phase specific

67
Methotrexate Structurally related to folic acid Enters cells by using the reduced folate carrier and the folate receptor protein Requires polyglutamation by the enzyme folylpolyglutamate synthase (FPGS) for its cytotoxic activity It inhibits DHFR and thus inhibits DNA synthesis –Inhibition of de novo thymidylate synthesis –Inhibition of de novo purine synthesis Incorporation of dUTP into DNA resulting in inhibition of DNA synthesis and function
 for its cytotoxic activity It inhibits DHFR and thus inhibits DNA synthesis –Inhibition of de novo thymidylate synthesis –Inhibition of de novo purine synthesis Incorporation of dUTP into DNA resulting in inhibition of DNA synthesis and function")
68
Pyrimidine analogs

69
Mechanism of action They inhibit the conversion of orotic acid to uidine (UMP) resulting in impaired nucleic acid synthesis Can be incorporated in DNA or RNA and cause point mutations thus disrupting flow of genetic information resulting in cell death 5FU can also act as a substrate for thymidilate synthase (TS) and cause inhibition of thymidine synthesis
 resulting in impaired nucleic acid synthesis Can be incorporated in DNA or RNA and cause point mutations thus disrupting flow of genetic information resulting in cell death 5FU can also act as a substrate for thymidilate synthase (TS) and cause inhibition of thymidine synthesis")
70
Mechanism of action Ara C when incorporated into DNA can interfere with DNA replication Gemcitabine, once phosphorylated, directly inhibits ribonucleotide reductase and inhibits do novo nucleotide production Gemcitabine may also be incorporated into DNA and interfere with DNA replication

71
Purine analogues

72
6 thioguanine (6TG) 6-mercaptopurine (6MP) Fludarabine 2deoxycoformycin (pentostatin) 2 chlorodeoxyadenosine (2-CdA)
 6-mercaptopurine (6MP) Fludarabine 2deoxycoformycin (pentostatin) 2 chlorodeoxyadenosine (2-CdA)")
73
Mechanism of action 6-thioguanine (6TG) and 6-mecaptopurine (6MP) –These agents are substrates for the enzyme hypxanthine guanine phosphoribosyl transferase and are converted to thioinosine. Thioinosine inhibits synthesis of adenine and guanine, thus interfering with DNA synthesis –thioinosine inhibits also the initial conversion of phophoribosyl pyrophosphate + glutamine to ribosylamine-5- phosphate (rate limiting initial step in purine biosynthesis)
.")
74
Mechanism of action Fludarabine –Fludarabine metabolites inhibit DNA polymerase, DNA primase and ribonulceotide reductase –Is incorporated into DNA and RNA 2deoxycoformycin –It inhibits adenosine deaminase leading to accumulation of intracellular adenosine and deoxyadenosine nucleotides, which blocks DNA synthesis by inhibiting ribonucleotide reductase 2 chlorodeoxyadenosine –An adenosine deaminase resistant purine analog that induces DNA strand breaks –It also induces NAD and ATP depletion and apoptosis

75
Clinical characteristics of antimetabolites Methotrexate: –acute lymphoblastic leukemia, osteosarcoma, lymphoma, carcinoma of the breast, ovary, bladder, head and neck, psoriasis, and rheumatoid arthritis 5 fluorouracil (5FU) –breast and colon cancer, esophageal cancer, squamous cell carcinoma of the head and neck Cytosine arabinoside (ara C): –acute myeloid leukemia and lymphoma Gemcitabine: –pancreatic cancer, non-small cell lung cancer
 –breast and colon cancer, esophageal cancer, squamous cell carcinoma of the head and neck Cytosine arabinoside (ara C): –acute myeloid leukemia and lymphoma Gemcitabine: –pancreatic cancer, non-small cell lung cancer")
76
Clinical characteristics of antimetabolites 6-thioguanine (6TG)/6-mecaptopurine (6MP): –acute lymphoblastic leukemia Fludarabine: –chronic lymphocytic leukemia, low grade lymphoma 2deoxycoformycin: –hairy cell leukemia, T- and B-cell lymphomas 2 chlorodeoxyadenosine: –hairy cell leukemia, chronic lymphocytic leukemia, low grade lymphoma
/6-mecaptopurine (6MP): –acute lymphoblastic leukemia Fludarabine: –chronic lymphocytic leukemia, low grade lymphoma 2deoxycoformycin: –hairy cell leukemia, T- and B-cell lymphomas 2 chlorodeoxyadenosine: –hairy cell leukemia, chronic lymphocytic leukemia, low grade lymphoma")
77
Toxicities of antimetabolites Methotrexate- –myelosuppression, hemorrhagic cystitis 5 fluorouracil (5FU) –mucositis, diarrhea Cytosine arabinoside (ara C), –myelosuppression, cerebellar dysfunction Gemcitabine, –myelosuppression, flu-like syndrome
 –mucositis, diarrhea Cytosine arabinoside (ara C), –myelosuppression, cerebellar dysfunction Gemcitabine, –myelosuppression, flu-like syndrome")
78
Toxicities of antimetabolites 6-thioguanine (6TG)/6-mecaptopurine (6MP) –Myelosuppression Fludarabine –myelosuppression, CD4 T cell depletion, peripheral neuropathy 2deoxycoformycin –myelosuppression 2 chlorodeoxyadenosine –myelosuppression, especially thrombocytopenia
/6-mecaptopurine (6MP) –Myelosuppression Fludarabine –myelosuppression, CD4 T cell depletion, peripheral neuropathy 2deoxycoformycin –myelosuppression 2 chlorodeoxyadenosine –myelosuppression, especially thrombocytopenia")
Similar presentations









 Carcinoembryonic.>")

. C C ancers most commonly occur in: breast (♀) ->")










