Download presentation
Presentation is loading. Please wait.

1
Structure prediction: Ab-initio Lecture 9 Structural Bioinformatics Dr. Avraham Samson 81-871 Let’s think!

2
Levinthal's paradox In 1969, Cyrus Levinthal noted that, because of the very large number of degrees of freedom in a polypeptide chain, the molecule has an astronomical number of possible conformations. For example, a polypeptide of 100 residues will have 99 peptide bonds, and therefore 198 different phi and psi bond angles. If each of these bond angles can be in one of three stable conformations, the protein may misfold into a maximum of 3 198 (~10 100 ) different conformations. Therefore, a polypeptide would require a time longer than the age of the universe to arrive at its correct native conformation. This is true even if conformations are sampled at rapid (picosecond) rates. The "paradox" is that most small proteins fold spontaneously on a millisecond or even microsecond time scale.Cyrus Levinthaldegrees of freedompolypeptide chainresiduespeptide bondsphi and psipicosecond
 different conformations. Therefore, a polypeptide would require a time longer than the age of the universe to arrive at its correct native conformation. This is true even if conformations are sampled at rapid (picosecond) rates. The paradox is that most small proteins fold spontaneously on a millisecond or even microsecond time scale.Cyrus Levinthaldegrees of freedompolypeptide chainresiduespeptide bondsphi and psipicosecond.")
3
3 Protein Structure Prediction Two main categories of protein structure prediction methods: – Homology modeling (class of last week!) – Ab-initio methods (class of today!) Methods can also be characterized: – Based on physical principles (simulations) – Based on statistics derived from known structures (knowledge-based)
 – Ab-initio methods (class of today!) Methods can also be characterized: – Based on physical principles (simulations) – Based on statistics derived from known structures (knowledge-based)")
4
4 Secondary Structure Prediction Methods attempt to decide which type of secondary structure (helix, strand or coil) each amino acid in a protein sequence is likely to adopt. The based methods are currently able to achieve success rates of over 75% based on sequence profiles.

5
5 Folding Simulations Accurate folding simulations will allow us to predict the structure of any protein. However, this approach is impractical due to limitations of computing power. Our understanding of the principles of protein folding are far short of the level needed to achieve this.

6
6 Homology Modeling Sometimes referred to as “Comparative modeling” The most reliable technique for predicting protein structure Comparing the sequence of the new protein with the sequences of proteins of known structure – Strong similarity (% identity, % similarity, alignment) – No strong similarities comparative modeling cannot be used. Similar sequences Almost identical structures

7
7 Predicting Small Conformational Changes Even between very similar proteins, there are differences. Some of these differences might be functionally important (different binding loop conformations) Predicting what the effects of these small structural changes is the real challenge in modeling Native fold of a protein can be found by finding the conformation of the protein which has the lowest energy as defined by a suitable potential energy function.
 Predicting what the effects of these small structural changes is the real challenge in modeling Native fold of a protein can be found by finding the conformation of the protein which has the lowest energy as defined by a suitable potential energy function..")
8
8 Ab initio Prediction Ab initio (i.e. ‘ from scratch ’ ) Use only the information in the target sequence itself Two branches – Knowledge-based methods Predict structure by applying statistical rules Rules: observations made on known protein structures – Simulation methods Predict structures by applying physical parameters (Van-der- Waals, dipole-dipole, etc)
 Use only the information in the target sequence itself Two branches – Knowledge-based methods Predict structure by applying statistical rules Rules: observations made on known protein structures – Simulation methods Predict structures by applying physical parameters (Van-der- Waals, dipole-dipole, etc).")
9
9 Simulation Methods Most ambitious approach Simulate the protein-folding process using basic physics Only useful for short peptides and small molecules Very useful for predicting unknown loop conformations as part of homology modeling

10
10 Energy Function The exact form of this energy function is as yet unknown It is reasonable to assume that it would incorporate terms pertaining to the types of interactions observed in protein structures – Hydrogen bonding – Van der Waals effects Find a potential function Construct an algorithm capable of finding the global minimum of this function

11
11 Searching Conformational Space Consider a protein chain of N residues The size of its conformational space is roughly 10 N states. 10 main chain torsion angle triples for each residue Not consider the additional conformational space provided by the side chain torsion yet.

12
12 How to Find Global Energy Minimum Efficiently Clearly proteins do not fold by searching their entire conformational space (Levinthal’s paradox) Proteins fold by means of a folding pathway encoded in the protein sequence ? Short-chain segments (5-7 residues) could quite easily locate their global minimum. Location of the native fold is driven by the folding of such short fragments ?
 could quite easily locate their global minimum. Location of the native fold is driven by the folding of such short fragments .")
13
13 One Subtle Point The native conformation need not necessarily correspond to the global minimum of free energy.

14
14 Secondary Structure Prediction Although predicting just the secondary structure of a protein is a long way from predicting its tertiary structure, information on the locations of helices and strands in a protein can provide useful insights as to its possible overall fold. It is also worth noting that the origins of the protein structure prediction field lie in this area

15
15 Intrinsic Propensities for Secondary Structure Formation Are some residues more likely to form -helices or -strands than others? Yes – Ex. proline residues are not often found in -helices 1974, statistical analysis of 15 proteins with known 3-D structures For each of the 20 amino acids, calculate the probability of finding any residue in -helices and in -strands Also calculate the probability of finding any residue in - helices and in -strands

16
16 Example (Chou and Fasman, 1974) Suppose there was a total of 2000 residues in their 15 protein data set Total number of residues2000 Number of alanines100 Number of helical residues500 Number of alanines in helices50 We would calculate the propensity of alanine for helix formation as follows: P(Ala in Helix) = 50/500 = 0.1 P(Ala) = 100/2000 = 0.05 Helix propensity (PA) of Ala = P(Ala in Helix)/P(Ala) = 0.1/0.05 = 2
 Suppose there was a total of 2000 residues in their 15 protein data set Total number of residues2000 Number of alanines100 Number of helical residues500 Number of alanines in helices50 We would calculate the propensity of alanine for helix formation as follows: P(Ala in Helix) = 50/500 = 0.1 P(Ala) = 100/2000 = 0.05 Helix propensity (PA) of Ala = P(Ala in Helix)/P(Ala) = 0.1/0.05 = 2")
17
AVVTW...GTTWVR ab-initio prediction Prediction from sequence using first principles

18
Ab-initio prediction “In theory”, we should be able to build native structures from first principles using sequence information and molecular dynamics simulations: “Ab-initio prediction of structure” – Simulation of the villin head piece (36-residues). (Pande et al.) http://www.youtube.com/watch?v=1eSwDK ZQpok&feature=related http://www.youtube.com/watch?NR=1&v= meNEUTn9Atg&feature=endscreen
 v=1eSwDK ZQpok&feature=related NR=1&v= meNEUTn9Atg&feature=endscreen.")
19
... the bad news... It is not possible to span simulations to the “seconds” range Simulations are limited to small systems and fast folding/unfolding events in known structures – steered dynamics – biased molecular dynamics Simplified systems

20
typical shortcuts Reduce conformational space – 1,2 atoms per residue – fixed lattices Statistic force-fields obtained from known structures – Average distances between residues – Interactions Use building blocks: 3-9 residues from PDB structures

21
“lattice” folding (2D) Self-avoidance is easily monitored! Energy is easily calculated
 Self-avoidance is easily monitored! Energy is easily calculated")
22
Example PROSA potential Total Hydrophobic C -C http://lore.came.sbg.ac.at:8080/CAME/CAME_EXTERN/ProsaII/index_html Very stable Low stability

23
Some protein from E.coli predicted at 7.6 Å (CASP3, H.Scheraga) Results from ab-initio Average error 5 Å - 10 Å Average error 5 Å - 10 Å Long simulations Long simulations
 Results from ab-initio Average error 5 Å - 10 Å Average error 5 Å - 10 Å Long simulations Long simulations")
24
Ab initio PDB “loops” in homology modeling

25
Final test The model must justify experimental data (i.e. differences between unknown sequence and templates) and be useful to understand function.
 and be useful to understand function..")
26
Rosetta energy function Residue environment (solvation) Residue pair interaction (electrostatic, disulfides) Steric repulsion Radius of gyration (vdw attraction, solvation) Cb density (solvation, correction for excluded volume) SS pairing (hydrogen bonding) Strand arrangement into sheet Helix-strand packing
 Residue pair interaction (electrostatic, disulfides) Steric repulsion Radius of gyration (vdw attraction, solvation) Cb density (solvation, correction for excluded volume) SS pairing (hydrogen bonding) Strand arrangement into sheet Helix-strand packing")
27
Protein Structure Prediction using ROSETTA

28
Worldwide distributed computing

29
Ab Initio Methods Ab initio: “From the beginning”. Assumption 1: All the information about the structure of a protein is contained in its sequence of amino acids. Assumption 2: The structure that a (globular) protein folds into is the structure with the lowest free energy. Finding native-like conformations require: - A scoring function (potential). - A search strategy.
 protein folds into is the structure with the lowest free energy. Finding native-like conformations require: - A scoring function (potential). - A search strategy..")
30
Rosetta The scoring function is a model generated using various contributions. It has a sequence dependent part (including for example a term for hydrophobic burial), and a sequence independent part (including for example a term for strand- strand packing). The search is carried out using simulated annealing. The move set is defined by a fragment library for each three and nine residue segment of the chain. The fragments are extracted from observed structures in the PDB.
, and a sequence independent part (including for example a term for strand- strand packing). The search is carried out using simulated annealing. The move set is defined by a fragment library for each three and nine residue segment of the chain. The fragments are extracted from observed structures in the PDB..")
31
The Rosetta Scoring Function

32
Hydrophobic Burial

33
Residue Pair Interaction

34
The Sequence Independent Term vector representation

35
Strand Packing – Helps! Estimated distribution

36
Sheer Angles – Help not!

37
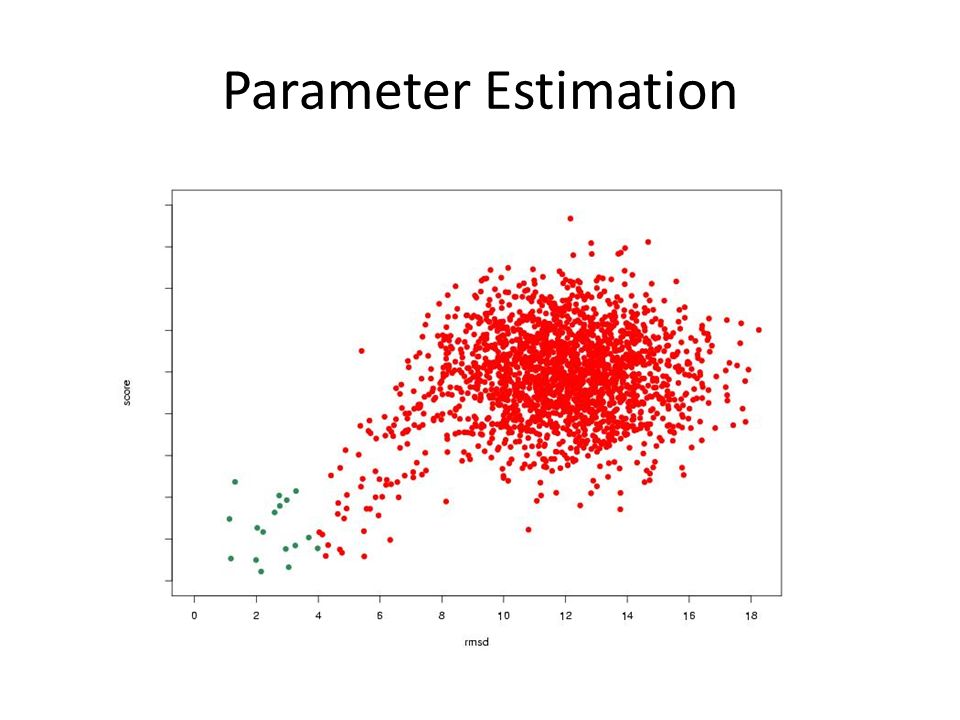
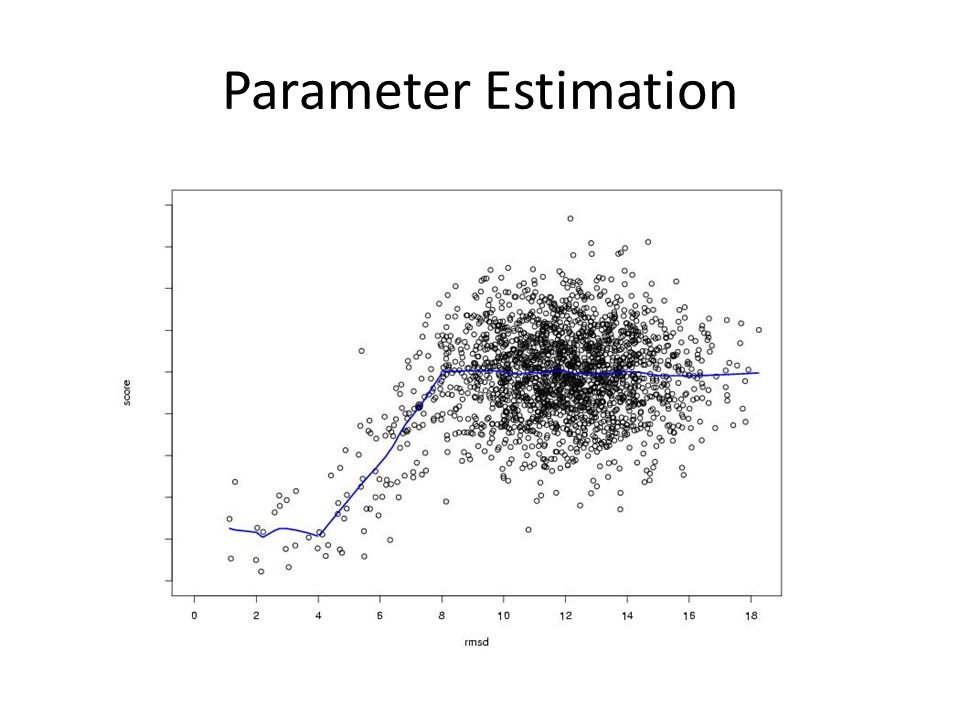
Parameter Estimation

41
Fragment Selection

43
Validation Data Set

44
CASP3 Protocol Construct a multiple sequence alignment from -blast. Edit the multiple sequence alignment. Identify the ab initio targets from the sequence. Search the literature for biological and functional information. Generate 1200 structures, each the result of 100,000 cycles. Analyze the top 50 or so structures by an all-atom scoring function (also using clustering data). Rank the top 5 structures according to protein-like appearance and/or expectations from the literature.
. Rank the top 5 structures according to protein-like appearance and/or expectations from the literature..")
45
CASP3 Predictions

46
Monte Carlo (Random Sampling) Randomly (or pseudorandomly) pick a configuration and evaluate its energy. If acceptably low, store result. If not, move a distance away from that point as a function of the energy (Metropolis criterion, a.k.a. simulated annealing) and evaluate again When some convergence threshold or time limit is met, stop and return stored results. http://www.chemistryexplained.com/images/c hfa_03_img0571.jpg Why is Rosetta so fast?
 and evaluate again When some convergence threshold or time limit is met, stop and return stored results. hfa_03_img0571.jpg Why is Rosetta so fast .")
47
What have we learned? Can tackle sampling today Forcefields sufficient? Folding to the native state folding rate prediction Role of water – Explicit solvent not crucial to rate determination? – Compare to explicit solvent simulation Universal mechanism of folding? – Maybe no universal mechanism: all proteins could be different?

Similar presentations





![Structure Prediction. Tertiary protein structure: protein folding Three main approaches: [1] experimental determination (X-ray crystallography, NMR) [2]](/15/4859888/big_thumb.jpg "Structure Prediction. Tertiary protein structure: protein folding Three main approaches: [1] experimental determination (X-ray crystallography, NMR) [2]>")

![Structure Prediction. Tertiary protein structure: protein folding Three main approaches: [1] experimental determination (X-ray crystallography, NMR) [2]](/16/4993760/big_thumb.jpg "Structure Prediction. Tertiary protein structure: protein folding Three main approaches: [1] experimental determination (X-ray crystallography, NMR) [2]>")



![. Protein Structure Prediction [Based on Structural Bioinformatics, section VII]](/16/5116302/big_thumb.jpg ". Protein Structure Prediction [Based on Structural Bioinformatics, section VII]>")
 Today: Why bother about proteins/prediction Concepts of molecular.>")
![Protein structure determination & prediction. Tertiary protein structure: protein folding Three main approaches: [1] experimental determination (X-ray.](/16/5177546/big_thumb.jpg "Protein structure determination & prediction. Tertiary protein structure: protein folding Three main approaches: [1] experimental determination (X-ray.>")







