Download presentation
Presentation is loading. Please wait.

1
Homology Modeling David Shiuan Department of Life Science and Institute of Biotechnology National Dong Hwa University

2
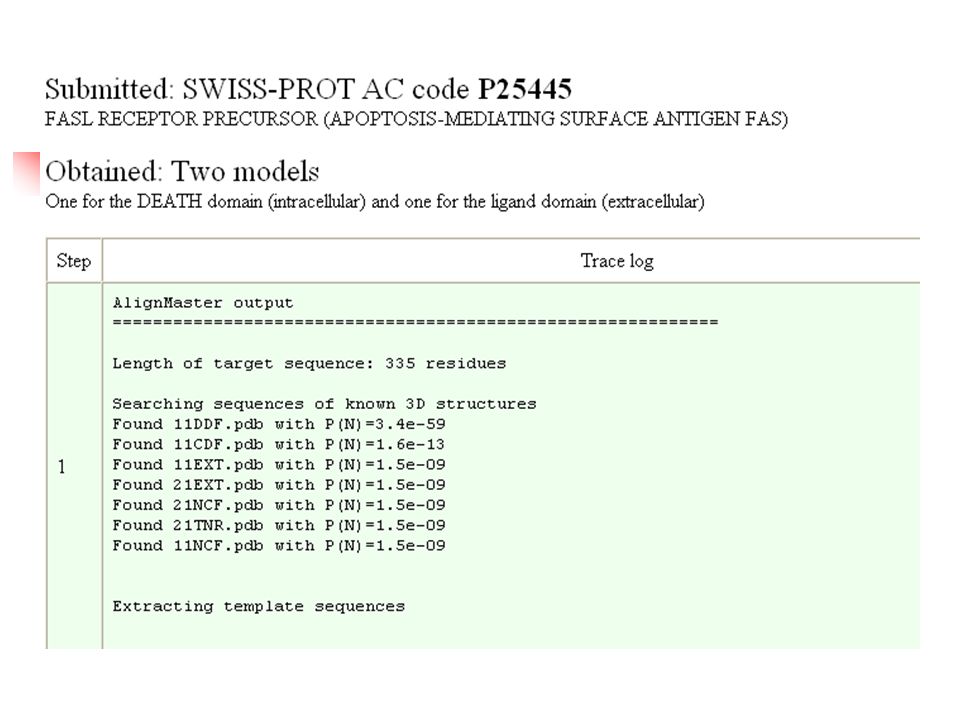
Why Modeling ? X-ray diffraction electron diffraction map electron density map Missing chains and residues in PDB structures No structure available

3
Erwin Schrodinger

4
John Pople 1964 Noble Computation Chemistry Walter Kohn 1960 Noble Density- Function theory

5
Polypeptide Chain

6
Structural Models are a unique source of information

7
The first solved protein crystal structure was of Sperm Whale myoglobin determined by Max Perutz and Sir John Cowdery Kendrew in 1958. They were awarded the Nobel Prize in Chemistry in 1962proteinSperm WhalemyoglobinMax PerutzSir John Cowdery KendrewNobel Prize in Chemistry

8
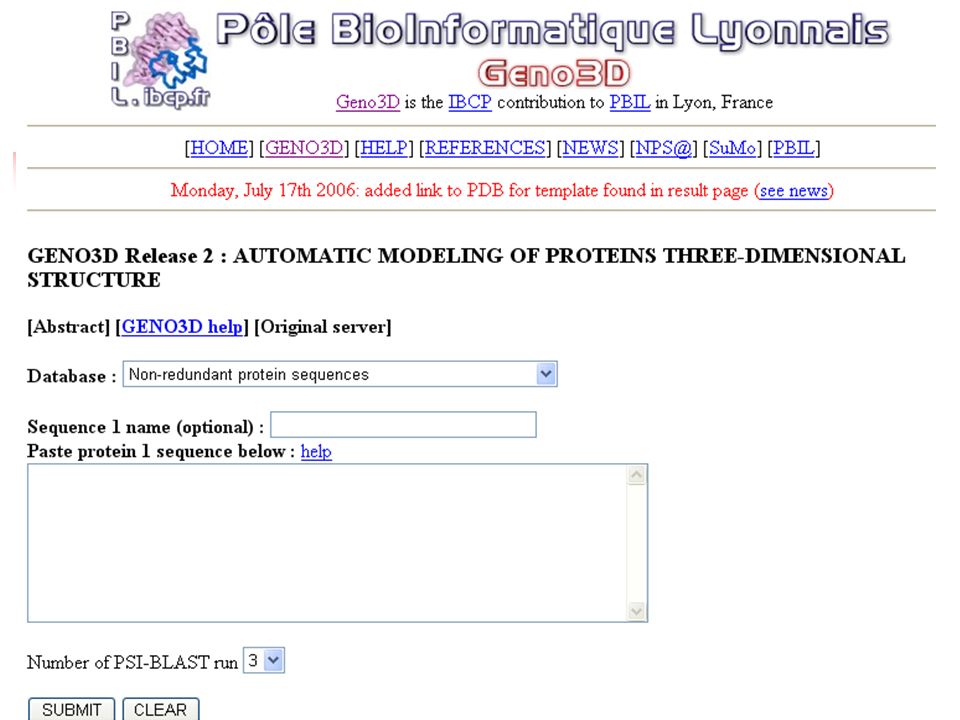
Modeling – Prediction of 3D Structures Homology Modeling Structures of similar molecules available Threading Prediction-based threading detecting the fold type and aligning a protein of unknown structure and a protein of known structure for low levels of sequence identity ( < 25%) Ab initio predicts the structure of proteins from the sequence and using molecular energy calculations (Schrodinger equation), do not use experimental parameters.
 Ab initio predicts the structure of proteins from the sequence and using molecular energy calculations (Schrodinger equation), do not use experimental parameters.")
12
Threading, A new approach to protein fold recognition. Nature 358 (1992 ) 86-89 An alternative strategy of recognizing known motifs or folds in sequences looks promising Threading is an approach to fold recognition which used a detailed 3-D representation of protein structure. The idea was to physically "thread" a sequence of amino acid side chains onto a backbone structure (a fold) and to evaluate this proposed 3-D structure using a set of pair potentials and (importantly) a separate solvation potential.
 An alternative strategy of recognizing known motifs or folds in sequences looks promising Threading is an approach to fold recognition which used a detailed 3-D representation of protein structure. The idea was to physically thread a sequence of amino acid side chains onto a backbone structure (a fold) and to evaluate this proposed 3-D structure using a set of pair potentials and (importantly) a separate solvation potential..")
18
View saccharide with JMol-Applet Chemis3D-Applet

19
Bystroff C & Shao Y. (2002). Fully automated ab initio protein structure prediction using I- SITES, HMMSTR and ROSETTA. Bioinformatics 18 Suppl 1, S54-61. Ab initio Structure Prediction

20
Comparative Protein Modelling Proteins with high sequence similarity is reflected by distinct structure similarity Comparative protein modelling (Homology Modeling) is presently the most reliable method. Comparative model building consist of the extrapolation of the structure for a new (target) sequence from the known 3D-structure of related family members (templates).
 sequence from the known 3D-structure of related family members (templates)..")
21
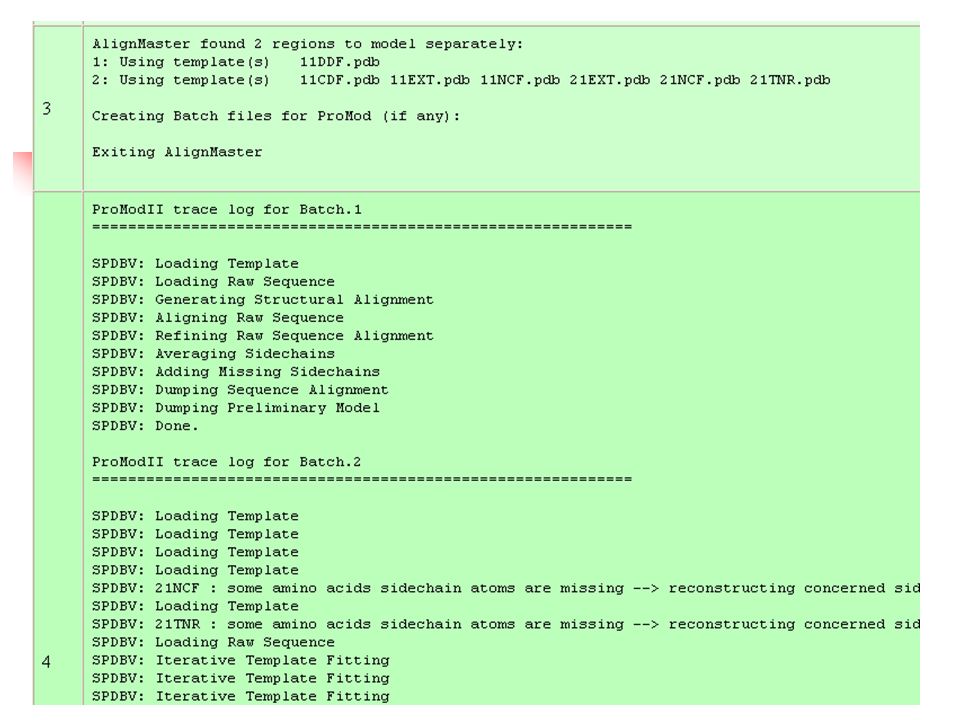
Building The Model 1. Framework construction By averaging the position of each atom in the target sequence, based on the location of the corresponding atoms in the template

22
Building The Model 2. Building non-conserved loops Although most of the known 3D-structures available share no overall similarity with the template, there may be similarities in the loop regions, and these can be inserted as loop structure in the new protein model

23
Building The Model 3. Completing the backbone Since the loop building only adds C atoms, the backbone carbonyl and nitrogens must be completed in these regions. This step can be performed by using a library of pentapeptide backbone fragments derived from the PDB entries

24
Building The Model 4. Adding side chains For many of the protein side chains there is no structural information available in the templates. These cannot therefore be built during the framework generation and must be added later

25
Building The Model 5. Model refinement Idealisation of bond geometry and removal of unfavourable non-bonded contacts can be performed by energy minimisation with force fields such as CHARMM, AMBER or GROMOS.

26
How to Superimpose Two Proteins Open the PDB file 11MUP11MUP Open the PDB file 21OBP21OBP Color by secondary structure Use the "Iterative Magic Fit" or the“Improve Fit" item of the "Tools" menu

28
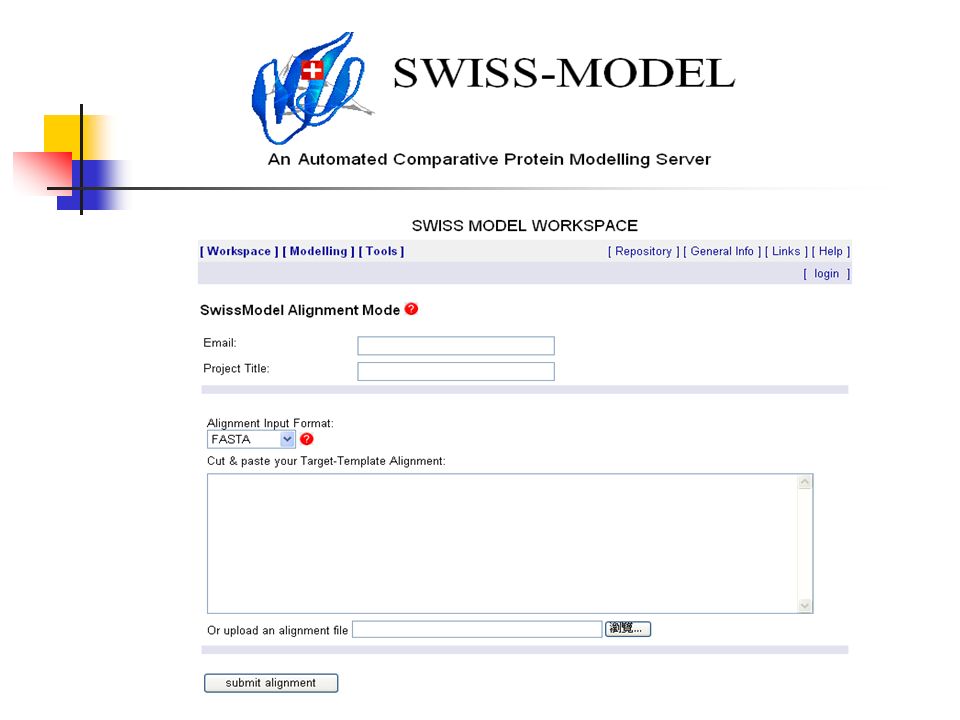
How SWISS-MODEL works

33
Probabilities of SWISS-MODEL accuracy for target-template identity classes

35
224 aa

43
We have identified three new families of insulin homologs in C. elegans. Comparative protein modelling remarkably confirms these predictions Example/Swiss Model: Insulin-likeInsulin-like growth factors in C. elegans.

Similar presentations





















![Structure Prediction. Tertiary protein structure: protein folding Three main approaches: [1] experimental determination (X-ray crystallography, NMR) [2]](/15/4859888/big_thumb.jpg "Structure Prediction. Tertiary protein structure: protein folding Three main approaches: [1] experimental determination (X-ray crystallography, NMR) [2]>")


![Protein structure determination. Tertiary protein structure: protein folding Three main approaches: [1] experimental determination (X-ray crystallography,](/16/4956805/big_thumb.jpg "Protein structure determination. Tertiary protein structure: protein folding Three main approaches: [1] experimental determination (X-ray crystallography,>")
 Protein Structure Prediction 3-Dimensional Structure.>")

![Structure Prediction. Tertiary protein structure: protein folding Three main approaches: [1] experimental determination (X-ray crystallography, NMR) [2]](/16/4993760/big_thumb.jpg "Structure Prediction. Tertiary protein structure: protein folding Three main approaches: [1] experimental determination (X-ray crystallography, NMR) [2]>")

>")
![. Protein Structure Prediction [Based on Structural Bioinformatics, section VII]](/16/5116302/big_thumb.jpg ". Protein Structure Prediction [Based on Structural Bioinformatics, section VII]>")


