Download presentation
Presentation is loading. Please wait.

1
Transient Protein-Protein Interactions (TPPI)
Presented By: Muhammad Shoaib Amjad 11 arid 3758 PhD Botany 1st Semester

2
Contents Protein-protein interactions Transient protein interaction
Transient protein and drug interaction Experimental techniques used for detecting TPPIs List of databases Summary References

3
Protein-Protein Interaction (PPI)
Interaction of two proteins Play an essential role in the proper functioning of living cells The forces responsible for these interactions include Electrostatic forces Hydrogen bonds Van der waals forces Hydrophobic effects Protein–protein interactions occur when two or more proteins bind together The understanding of these interactions will provide the clues to their biological function

4
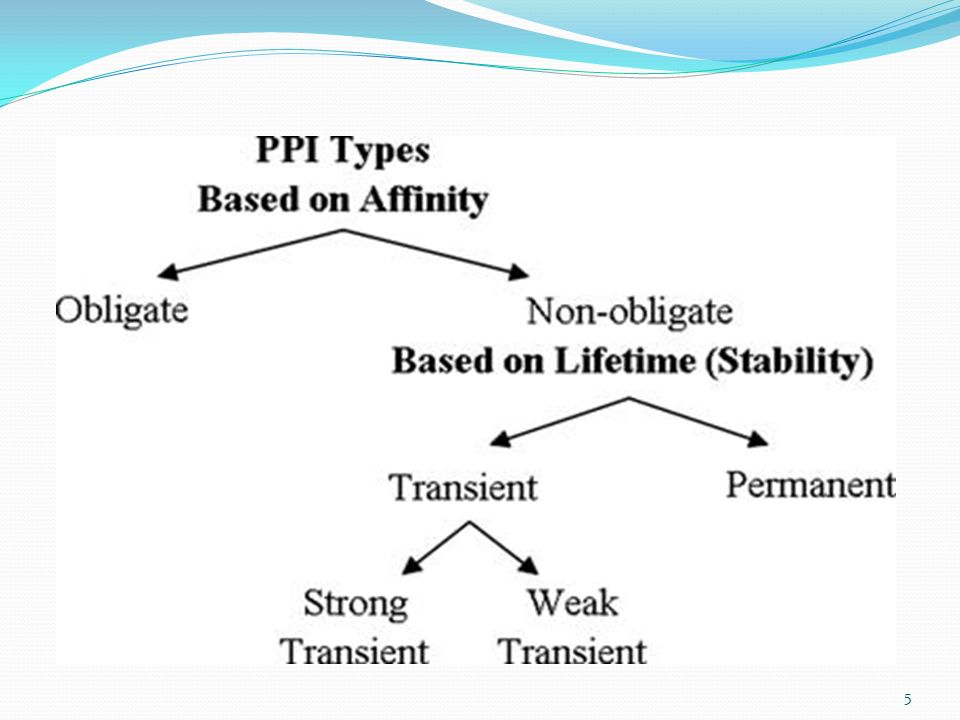
Types of Potien-Protien interactions
PPIs can be classified on the bases of Composition Homo and hetero-oligomeric complexes Affinity Non-obligate and obligate complexes Stability/Life time Transient and permanent

6
Transient and Permanent
Stable Irreversible Strong Long life Electrostatic force Example: α/β tubulin dimer and many enzyme-inhibitor complexes

7
Transiet protein-protein interactions (TPPI)
TPPIs are involved in many biological processes: Signal transduction Protein complexes or molecular machinery Protein carrier Protein modifications (phosphorylation) Hormone receptor binding Allostery of enzymes Inhibition of proteases Correction of misfold protein by chaperones TPPIs help to decipher the molecular mechanisms underlying the biological functions, and enhance the approaches for drug discovery For example, signals from the exterior of a cell are mediated to the inside of that cell by protein-protein interactions of the signalling molecules In protein complex, members are linked by non-covalent interactions, they often activate or inhibit other members. a protein may be carrying another protein, for example, from cytoplasm to nucleus or vice versa in the case of the nuclear pore importins, a type of protein that moves other protein molecules into the nucleus by binding to a specific recognition sequence protein kinase will add a phosphate to a target protein
 Hormone receptor binding. Allostery of enzymes. Inhibition of proteases. Correction of misfold protein by chaperones. TPPIs help to decipher the molecular mechanisms underlying the biological functions, and enhance the approaches for drug discovery. For example, signals from the exterior of a cell are mediated to the inside of that cell by protein-protein interactions of the signalling molecules. In protein complex, members are linked by non-covalent interactions, they often activate or inhibit other members. a protein may be carrying another protein, for example, from cytoplasm to nucleus or vice versa in the case of the nuclear pore importins, a type of protein that moves other protein molecules into the nucleus by binding to a specific recognition sequence. protein kinase will add a phosphate to a target protein.")
8
Transient protein and drug interaction
Transient interactions might be important in drug mechanisms in two ways: the drugs that act on TPPIs act transiently on their multiple targets A cancer-related example for the former type of drugs is nutlins

9
Transient protein and drug interaction
Nutlins MDM2 p53 Tumor suppressed

10
Experimental Techniques
Yeast two-hybrid screens Mass spectrometry Intracellular localization of proteins with fluorescence markers

11
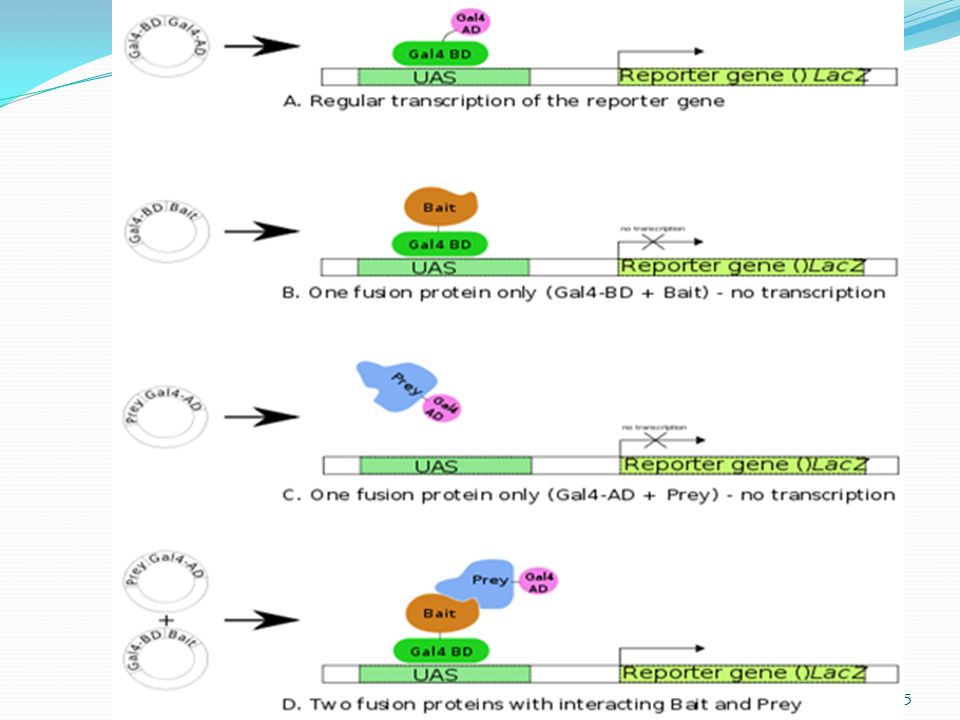
The Yeast Two-Hybrid System

12
Yeast Two-hybrid Researchers insert a gene in yeast for a "bait" protein alongside DNA for half of an "activator" protein. The other half of the activator DNA is then inserted alongside DNA for random "prey" proteins. The yeast cells are then grown up and the proteins are allowed to interact. If bait and prey proteins bind, the two halves of the activator protein be close enough to work together to turn on another yeast gene that turns the cell blue, signaling a match.

13
How does it work? Uses yeast as a model for eukaryotic protein interactions A library is screened or a protein is characterized using a bait construct Interactions are identified by the transcription of reporter genes Positives are selected using differential media

14
DNA-Binding Domain Bait Protein Prey Protein Transcription Activating Region DNA-Binding Site Reporter Gene

16
What is the yeast two-hybrid system used for?
Identifies novel protein-protein interactions Can identify protein cascades Identifies mutations that affect protein-protein binding Can identify interfering proteins in known interactions

17
Steps to Screen a Library
Create the Bait Plasmid Construct from the gene of interest and the DNA binding domain of Gal4 or LexA or other suitable domain Transform with the bait construct a yeast strain lacking the promoter for the reporter genes and select for transformed yeast Transform the yeast again with the library plasmids Select for interaction

18
Sequence analysis Isolate plasmid from yeast and transform E. coli
Purify plasmid from E. coli and sequence Blast sequence against database for known proteins or construct a possible protein sequence from the DNA sequence and compare to other proteins

19
Sample Plasmid From Golemis Lab Homepage

20
Reporter Genes LacZ reporter - Blue/White Screening
HIS3 reporter - Screen on His+ media LEU2 reporter - Screen on Leu+ media ADE2 reporter - Screen on Ade+ media URA3 reporter - Screen on Ura+ media

21
False Positives False positives are the largest problem with the yeast two-hybrid system Can be caused by: Non-specific binding of the prey Ability to induce transcription without interaction with the bait (Majority of false positives)
")
22
Elimination of False Positives
Sequence Analysis Plasmid Loss Assays Retransformation of both strain with bait plasmid and strain without bait plasmid Test for interaction with an unrelated protein as bait Two (or more) step selections
 step selections.")
23
Advantages Immediate availability of the cloned gene of the interacting protein Only a single plasmid construction is required Interactions are detected in vivo Weak, transient interactions can be detected Can accumulate a weak signal over time

24
Examples of Uses of the Yeast Two-Hybrid System
Identification of caspase substrates Interaction of Calmodulin and L-Isoaspartyl Methyltransferase Genetic characterization of mutations in E2F1 Peptide hormone-receptor interactions Pha-4 interactions in C. elegans

25
The Study of Protein-protein Interaction by Mass Spectrometry
Direct analysis of large protein complexes (DALPC). In the flow diagram, the rectangles represent a strong cation exchange (SCX) and a reversed-phase (RP) liquid chromatography column. Typically, a denatured protein complex is digested with trypsin. The acidified peptide mixture is loaded onto the SCX column. A discrete fraction of peptides is displaced from the SCX column to the RP column. This fraction is eluted from the RP column into the mass spectrometer. This iterative process is repeated, obtaining the fragmentation patterns of peptides in the original peptide mixture. The program SEQUEST is used to correlate the tandem mass spectra of fragmented peptides to amino acid sequences using nucleotide databases 6 . The filtered outputs from the program are used to identify the proteins in the original protein complex.
. In the. flow diagram, the rectangles represent a strong cation exchange. (SCX) and a reversed-phase (RP) liquid chromatography column. Typically, a denatured protein complex is digested with trypsin. The. acidified peptide mixture is loaded onto the SCX column. A discrete. fraction of peptides is displaced from the SCX column to the RP. column. This fraction is eluted from the RP column into the mass. spectrometer. This iterative process is repeated, obtaining the. fragmentation patterns of peptides in the original peptide mixture. The program SEQUEST is used to correlate the tandem mass. spectra of fragmented peptides to amino acid sequences using. nucleotide databases The filtered outputs from the program are. used to identify the proteins in the original protein complex.")
26
Databases

28
ELM is a computational biology resource for investigating candidate functional sites in eukarytic proteins. Functional sites which fit to the description "linear motif" are currently specified as patterns using Regular Expression rules. To improve the predictive power, context-based rules and logical filters are being developed and applied to reduce the amount of false positives. The current version of the ELM server provides core functionality including filtering by cell compartment, phylogeny, globular domain clash (using the SMART/Pfam databases) and structure. In addition, both the known ELM instances and any positionally conserved matches in sequences similar to ELM instance sequences are identified and displayed (seeELM instance mapper). Although the ELM resource contains a large collection of functional site motifs, the current set of motifs is not exhaustive.
 and structure. In addition, both the known ELM instances and any positionally conserved matches in sequences similar to ELM instance sequences are identified and displayed (seeELM instance mapper). Although the ELM resource contains a large collection of functional site motifs, the current set of motifs is not exhaustive.")
29
Our group is affiliated with the Departments of Environmental Health and Biomedical Engineering,University of Cincinnati College of Medicine, as well as with the Division of Biomedical Informatics,Children's Hospital Research Foundation (see links below). Our research in the fields ofbioinformatics and computational biology is driven by both methodological developments and applications, spanning a wide range of projects that deal with various data streams and problems arising in the context of genomics, proteomics and biomedical research. Some examples includeprotein structure and function prediction, protein-protein interactions and the recognition of functional sites in proteins, gene (genome) annotation, and the identification of predictive fingerprints of disease states, especially in the context of large SNP genotyping. We rely heavily onpattern recognition and machine learning approaches to develop improved and/or tailored methods for the above applications. In collaboration with biologists, biochemists and physicians, we are applying standard and our own methods to a particular problem at hand.
 annotation, and the identification of predictive fingerprints of disease states, especially in the context of large SNP genotyping. We rely heavily onpattern recognition and machine learning approaches to develop improved and/or tailored methods for the above applications. In collaboration with biologists, biochemists and physicians, we are applying standard and our own methods to a particular problem at hand..")
30
The AMS tool allows for identification of PTM (post-translational modification) sites in proteins. The local sequence preferences of short segments around PTM residues are described here as SVM based LFM (linear functional motifs). We train support vector machine (SVM) for each type of PTM separately on proteins of the Swiss-Prot database (version 42.0) using two datasets of short sequence segments: positives and negatives. The set of positives is created from experimentally annotated sequence segments (9-amino acids long) from proteins known to include PTM site. The dataset of negative cases is build from short sequence segments not annotated in Swiss-Prot database to include any type of PTM site. These two datasets are then projected as sets of points into a single multidimensional space. For this purpose we used generic projection methods of short segments and additional combinations of these. The SVM constructs the separation plane in the multidimensional space between the sets of positives and negatives. The identification of PTM sites in a query protein is performed as follows. The query protein is divided into overlapping sequence segments. Two search procedures can be used to annotate these segments: identity scan or SVM classification. The first method identifies those segments that are identical in terms of sequence to any positive instance from the database. The second method uses SVM to classify the query protein’s segments into two groups: potential PTM sites and negatives. The list of PTM sites found in the protein by any projection method is then returned to the user. The performance of SVM models is described by the recall R and the precision P. The recall R value measures the percentage of correct predictions (the probability of correct prediction), whereas precision P gives the percentage of observed positives that are correctly predicted (the measure of the reliability of positive instances prediction). These measures of accuracy are calculated separately for each type of PTM using the leave-one-out procedure. The typical recall value is around 30%, and the precision P is over 70% for majority of PTM.
, whereas precision P gives the percentage of observed positives that are correctly predicted (the measure of the reliability of positive instances prediction). These measures of accuracy are calculated separately for each type of PTM using the leave-one-out procedure. The typical recall value is around 30%, and the precision P is over 70% for majority of PTM.")
31
Summary The components of transient complexes associate and dissociate rapidly while transiently interacting with each other to function dynamically in crucial regulatory and signaling pathways. The identification and analysis of these complexes have become more manageable with the emerging sensitive and high-resolution experimental techniques accompanied by the high-throughput computational methods.

32
As the coverage of these techniques increase, they can provide a good template to understand and design new transient complexes. An example for such advanced techniques is, PRISM, which uses available transient interactions as a template set and searches structural and evolutionary similarities between the template set and the target proteins to be predicted.

33
References James R. Perkins, I. Diboun, B.H. Dessailly, J. G. Lees and C. Orengo Transient Protein- Protein Interactions: Structural Functional And Network Properties. Cell: Saliha E. A. O., H. B. Engin, A. Gursoy and O. Keskin Transient Protein- Protein Interactions. Protein Interaction Designed And selection: 1-14 Costel C. D., K. Deinhardt, G. Zhang, H. S. Cardasis and M. V. C.A. Neubert Identifying transient protein–protein interactions in EphB2 signaling by blue native PAGE and mass spectrometry Proteomics J:11, 4514–4528 Lakshmipuram S. S., R. M. Bhaskara, J. Sharma and N. Srinivasan Roles of residues in the interface of transient protein-protein complexes before complexation. Scientific reports: 334.

34
THANK YOU

Similar presentations
















. Detecting protein function and protein-protein interactions from genome sequences.>")



