Download presentation
Presentation is loading. Please wait.

1
Congenital defects of female genital tract
MUDr. R. Malina, Ph.D.

2
Uterovaginal anomalies are associated with a high incidence of decreased fertility and multiple obstetric problems. These anomalies are caused by alterations in development or fusion of the müllerian ducts.

3
Uterovaginal anomalies are classified into three types:
dysgenesis, vertical or lateral fusion defects, and unusual configurations.

4
The identification of uterovaginal anomalies is important in the treatment of infertility and symptoms that arise from an obstructed or deformed reproductive tract. Correct diagnosis and classification of uterovaginal anomalies is needed to determine cases requiring interventional therapy

5
Background

6
Embryology The female reproductive system develops from the müllerian ducts, two ducts that originate in embryonal mesoderm lateral to each wolffian duct. The paired müllerian ducts grow in medial and caudal directions. The most cephalad parts of the ducts remain separate and form the fallopian tubes. The lower parts of the ducts fuse (lateral fusion). The midline septum disappears, leaving a single canal: the uterus and upper two-thirds of the vagina.
. The midline septum disappears, leaving a single canal: the uterus and upper two-thirds of the vagina.")
7
The lower third of the vagina is formed from the ascending sinovaginal bulb, which fuses with the lower müllerian system (vertical fusion) (Fig 1) Figure 1. Development of the uterovaginal canal. Two paired müllerian ducts (yellow lines) grow medially and caudad. Both ducts fuse in the midline to form the uterus and the upper two-thirds of vagina (lateral fusion). The lower third of vagina is formed by fusion of the ascending sinovaginal bulb (red lines) with the müllerian system (vertical fusion). The septum disappears, leaving a single uterovaginal canal.
 grow medially and caudad. Both ducts fuse in the midline to form the uterus and the upper two-thirds of vagina (lateral fusion). The lower third of vagina is formed by fusion of the ascending sinovaginal bulb (red lines) with the müllerian system (vertical fusion). The septum disappears, leaving a single uterovaginal canal.")
8
The close developmental relationship of the müllerian and wolffian ducts explains the frequent association of anomalies of the female genital system and urinary tract. Renal anomalies associated with uterovaginal anomalies include renal agenesis, ectopic kidney, cystic dysplasia, and a duplicated collecting system. The associated renal anomaly is ipsilateral to the abnormally developed müllerian duct, since both are dependent on adequate development of the mesonephric system.

9
Prevalence and Distribution
In the general population, the prevalence of uterine anomalies is 0.5% and of vaginal anomalies is 0.025%. According to Nahum et al, the distribution of uterine anomalies is 4% hypoplastic, aplastic, or solid; 34% septate; 39% bicornuate; 7% arcuate; 11% didelphic; and 5% unicornuate.

10
Figure 2. Diagram of a unicornuate uterus.
Unicornuate uterus results from complete or incomplete arrest of development of one müllerian duct (Fig 2). The other horn (rudimentary) could be absent in 35% of cases, solid noncavitary in 33%, cavitary but noncommunicating with the uterine cavity in 22%, and cavitary communicating with the uterine cavity in 10% (9). Figure 2. Diagram of a unicornuate uterus. Saleem S N Radiographics 2003;23:e13-e13 ©2003 by Radiological Society of North America
. The other horn (rudimentary) could be absent in 35% of cases, solid noncavitary in 33%, cavitary but noncommunicating with the uterine cavity in 22%, and cavitary communicating with the uterine cavity in 10% (9). Figure 2. Diagram of a unicornuate uterus. Saleem S N Radiographics 2003;23:e13-e13. ©2003 by Radiological Society of North America.")
11
Unicornuate uterus

13
Figure 3. Anatomy before (a) and after (b) removal of the rudimentary horn.
Unicornuate uterus (UU), rudimentary horn (RH). The pictures were taken during laparoscopic surgery. Before surgery (a) and after surgery (b) with the rudimentary horn removed.
, rudimentary horn (RH). The pictures were taken during laparoscopic surgery. Before surgery (a) and after surgery (b) with the rudimentary horn removed.")
14
Figure 3. Diagram of a didelphic uterus.
Didelphic uterus results from complete nonfusion of both müllerian ducts (Fig 3). Two uterine bodies and two cervices are present. A longitudinal or transverse vaginal septum may be associated with this anomaly Figure 3. Diagram of a didelphic uterus. Saleem S N Radiographics 2003;23:e13-e13 ©2003 by Radiological Society of North America
. Two uterine bodies and two cervices are present. A longitudinal or transverse vaginal septum may be associated with this anomaly. Figure 3. Diagram of a didelphic uterus. Saleem S N Radiographics 2003;23:e13-e13. ©2003 by Radiological Society of North America.")
15
Figure 4. Diagram of a bicornuate uterus.
Bicornuate uterus results from partial nonfusion of the müllerian ducts (Fig 4). Double uterine bodies and a single cervix are present. An arcuate uterus is considered a milder form of bicornuate uterus; it has a convex or flat external fundal contour and mild impression on the endometrial cavity. Bicornuate bicollis is a variant of bicornuate uterus in which the anomaly is combined with a muscular uterine septum that extends to the external os . Figure 4. Diagram of a bicornuate uterus. Saleem S N Radiographics 2003;23:e13-e13 ©2003 by Radiological Society of North America
. Double uterine bodies and a single cervix are present. An arcuate uterus is considered a milder form of bicornuate uterus; it has a convex or flat external fundal contour and mild impression on the endometrial cavity. Bicornuate bicollis is a variant of bicornuate uterus in which the anomaly is combined with a muscular uterine septum that extends to the external os . Figure 4. Diagram of a bicornuate uterus. Saleem S N Radiographics 2003;23:e13-e13. ©2003 by Radiological Society of North America.")
16
Bicornuate uterus picture Left uterine horn on left side of photo, right horn on right Right ovary (white structure) visible at bottom right Dark blue dye at bottom was injected during surgery to document open fallopian tubes A bicornuate uterus should not cause infertility, but is associated with increased risks for miscarriage and preterm birth. It can be diagnosed using high quality 3D ultrasound and can be suspected from the results of a hysterosalpingogram, HSG (dye test). A bicornuate uterus vs. a septate uterus can not be distinguished by the HSG alone or by hysteroscopy, but 3D ultrasound of the uterus or laparoscopy can make the distinction between them.
. A bicornuate uterus vs. a septate uterus can not be distinguished by the HSG alone or by hysteroscopy, but 3D ultrasound of the uterus or laparoscopy can make the distinction between them.")
17
Arcuate Uterus

19
Figure 5. Diagram of a septate uterus.
Septate uterus results from complete fusion of the müllerian ducts with failure of resorption of the central septum (Fig 5). The septum may be partial or complete and fibrous or muscular. Figure 5. Diagram of a septate uterus. Saleem S N Radiographics 2003;23:e13-e13 ©2003 by Radiological Society of North America
. The septum may be partial or complete and fibrous or muscular. Figure 5. Diagram of a septate uterus. Saleem S N Radiographics 2003;23:e13-e13. ©2003 by Radiological Society of North America.")
20
Absent Uterus

21
Di-ethyl-stilbestrol (DES)–exposed uterus results when the fetal uterus is exposed to the estrogen analog DES, which causes abnormal myometrial hypertrophy.

22
Classification of Uterovaginal Anomalies
Classification of subtypes of congenital abnormalities of the female reproductive system is important in the treatment of infertility and symptoms arising from obstruction or deformity Many classifications of uterine anomalies exist; for instance, the Buttram and Gibbons classification and the American Fertility Society (AFS) classification.
 classification.")
24
Class 1.—Dysgenesis of müllerian ducts.
This class includes agenesis or hypoplasia of the müllerian duct derivatives: the uterus and upper two-thirds of the vagina. The most common form is the Mayer-Rokitansky-Kuster-Hauser syndrome, which is combined agenesis of the uterus, cervix, and upper portion of the vagina.

25
Mayer-Rokitansky-Kuster-Hauser (MRKH) syndrome
consists of vaginal aplasia with other müllerian (ie, paramesonephric) duct abnormalities. Its penetrance varies, as does the involvement of other organ systems. Type I Mayer-Rokitansky-Kuster-Hauser syndrome is characterized by an isolated absence of the proximal two thirds of the vagina, whereas type II is marked by other malformations; these include vertebral, cardiac, urologic (upper tract), and otologic anomalies.[2]
 duct abnormalities. Its penetrance varies, as does the involvement of other organ systems. Type I Mayer-Rokitansky-Kuster-Hauser syndrome is characterized by an isolated absence of the proximal two thirds of the vagina, whereas. type II is marked by other malformations; these include vertebral, cardiac, urologic (upper tract), and otologic anomalies.[2]")
26
In both types, the extent of vaginal aplasia varies, ranging from virtually absent to virtually inconsequential. Mayer-Rokitansky-Kuster-Hauser syndrome usually remains undetected until the patient presents with primary amenorrhea despite normal female sexual development. Mayer-Rokitansky-Kuster-Hauser syndrome is the second most common cause of primary amenorrhea. Although this condition has psychologically devastating consequences, its physiological defects can be surgically treated. Following diagnosis, surgical intervention allows patients to have normal sexual function. Reproduction may be possible with assisted techniques.

27
Pathophysiology At approximately 5 weeks' gestation, the müllerian ducts stop developing. The skeleton, which is derived from the embryonic mesoderm, is vulnerable to developmental disturbances at this time. The uterus, cervix, and upper two thirds of the vagina form from the fused caudal ends of the müllerian ducts. Fallopian tubes develop from the unfused upper ends; the renal system simultaneously develops from the wolffian (ie, mesonephric) ducts. Ovarian function is preserved because the ovaries originate within the primitive ectoderm, independent of the mesonephros. Although Mayer-Rokitansky-Kuster-Hauser syndrome was previously thought to be a sporadic anomaly, familial cases support the hypothesis of a genetic etiology and are receiving increased attention. Although the precise gene has not yet been identified, this syndrome appears to be transmitted in an autosomal dominant fashion, with incomplete penetrance and variable expressivity.
 ducts. Ovarian function is preserved because the ovaries originate within the primitive ectoderm, independent of the mesonephros. Although Mayer-Rokitansky-Kuster-Hauser syndrome was previously thought to be a sporadic anomaly, familial cases support the hypothesis of a genetic etiology and are receiving increased attention. Although the precise gene has not yet been identified, this syndrome appears to be transmitted in an autosomal dominant fashion, with incomplete penetrance and variable expressivity.")
28
Epidemiology Frequency
United States The incidence of congenital absence of the vagina is 1 per female births. Mayer-Rokitansky-Kuster-Hauser syndrome is generally thought to be a sporadic condition, and female relatives of the patient apparently have no increased risk. However, familial clustering is reported with increasing frequency. Mortality/Morbidity Mayer-Rokitansky-Kuster-Hauser syndrome has psychological consequences, but its physiological defects are surgically treatable. Surgical correction permits normal sexual function and, possibly, reproduction with assisted techniques. Race Mayer-Rokitansky-Kuster-Hauser syndrome has no racial predisposition. Sex Mayer-Rokitansky-Kuster-Hauser syndrome only affects females. Age Mayer-Rokitansky-Kuster-Hauser syndrome is a congenital disorder that is present at birth. However, it may remain undiagnosed until adolescence or early adulthood.

29
History The following may be observed in patients with Mayer-Rokitansky-Kuster-Hauser (MRKH) syndrome: Primary amenorrhea and possible cyclic abdominal pain These symptoms are common in individuals with Mayer-Rokitansky-Kuster-Hauser syndrome. The patient undergoes puberty with normal thelarche and adrenarche; however, menses do not begin. Patients may report cyclic abdominal pain due to cyclic endometrial shedding without a patent drainage pathway. Because ovarian function is normal, patients experience all bodily changes associated with menstruation and puberty.

30
Infertility Patients who do not undergo evaluation for primary amenorrhea often seek clinical attention for infertility. However, patients rarely proceed to infertility evaluation without ever having had a menses due to Mayer-Rokitansky-Kuster-Hauser syndrome. Although the ovaries function normally, the fallopian tubes may be closed, and the uterus is often anomalous. Inability to have intercourse The degree of vaginal aplasia can vary from complete absence to a blind pouch. The more shallow the canal, the greater the likelihood of the patient having dyspareunia. Renal malformations Absence or ectopia of the kidneys is common. Diagnosis can lead to discovery of renal anomalies. Some patients present with a history of voiding difficulties, urinary incontinence, or recurrent urinary tract infections (UTIs). Vertebral anomalies: Skeletal findings range in severity and clinical importance. Scoliosis is the most common of the skeletal anomalies.
. Vertebral anomalies: Skeletal findings range in severity and clinical importance. Scoliosis is the most common of the skeletal anomalies.")
31
Physical Normal secondary female sexual characteristics are present after puberty. Height is normal. Speculum examination of the vagina may be impossible or difficult because of the degree of vaginal agenesis. The vulva, labia majora, labia minora, and clitoris are normal. A palpable sling of tissue may be present at the level of the peritoneal reflection.

32
Causes The cause of Mayer-Rokitansky-Kuster-Hauser syndrome is unknown, and no known gene is linked to this condition. A postulation is that the müllerian duct system ceases development during gestational days

33
Differential Diagnoses
5-Alpha-Reductase Deficiency Androgen Insensitivity Syndrome Congenital Adrenal Hyperplasia Hermaphroditism Müllerian-inhibiting substance deficiency Turner Syndrome

34
Class 2.—Disorders of vertical fusion.
These anomalies are due to failure of fusion of the müllerian system with the sinovaginal bulb. They include cervical dysgenesis and obstructive and nonobstructive transverse vaginal septa.

36
Vertical Fusion Defect
Transverse vaginal septum.—A transverse vaginal septum could be in a high, middle, or low position. It is more common in the upper vagina Transverse vaginal septum (class 2). Sagittal T2-weighted spin-echo image (4,000/98) shows a transverse septum in the middle of the vagina (arrow), causing dilatation of the proximal vagina (V) and uterus (U) (hematocolpos and hetmatometria).
. Sagittal T2-weighted spin-echo image (4,000/98) shows a transverse septum in the middle of the vagina (arrow), causing dilatation of the proximal vagina (V) and uterus (U) (hematocolpos and hetmatometria).")
37
Obstructed hymen. Obstructed hymen is also considered a vertical fusion defect. Sagittal MR images document the exact site of vaginal obstruction. Consequent dilatation of the proximal reproductive canal filled with obstructed menstrual blood products is usually noted (Fig 11).
.")
40
HYMEN normální

41
Extensive labial adhesion. Vaginal agenesis.
Not to be confused with imperforate hymen. Vaginal agenesis. Not to be confused with imperforate hymen.

43
Class 3: Lateral fusion defects
A. Symmetric nonobstructive form of lateral fusion defects Unicornuate uterus.— A unicornuate uterus is banana-shaped and slender, without the usual rounded fundal contour, and is usually laterally deviated. Cervical agenesis—No identifiable cervix in MR images. Hematometria may be seen if there is functioning endometrium in the uterine body Septate Uterus—The outer fundal contour is convex or flat or has a slight indentation less than 1 cm deep. The intercornual distance is within the normal range. The intercornual angle measures less than 60°. Bicornuate uterus.—Two uterine bodies and a single cervix are present in bicornuate uterus. The fundal cleft is greater than 1 cm in depth. The intercornual distance is increased (>4 cm) in bicornuate uteri. Didelphic uterus.—Two uterine bodies and two cervices are present in didelphic uterus.
 in bicornuate uteri. Didelphic uterus.—Two uterine bodies and two cervices are present in didelphic uterus.")
45
B. Asymmetric obstructive form of lateral fusion defects
Unicornuate uterus with a noncommunicating rudimentary horn.—An obstructed rudimentary horn with functioning endometrium may be distended by blood or blood products. Retrograde menstruation into the fallopian tube may lead to associated hematosalpinx Unilateral obstruction of a cavity of a double uterus.—This is a unique syndrome consisting of a didelphic obstructed hemivagina and ipsilateral renal agenesis

46
This is a unique syndrome consisting of a didelphic obstructed hemivagina and ipsilateral renal agenesis

48
Class 3.—Disorders of lateral fusion.
This class describes anomalies that result in a duplicated or partially duplicated reproductive tract. The disorders are due to impaired fusion and/or septal resorption of fusing müllerian ducts attempting to form the uterus, cervix, and upper vagina. It includes anomalies due to failure of fusion of the paired müllerian ducts (as in didelphic and bicornuate uteri) and failure of midline septum resorption after fusion (as in septate uterus).
 and failure of midline septum resorption after fusion (as in septate uterus).")
49
Disorders due to lateral fusion defects are further subclassified into
(a) the symmetric nonobstructive form seen in five types: unicornuate, bicornuate, didelphic, septate, and DES-related uteri
 the symmetric nonobstructive form seen in five types: unicornuate, bicornuate, didelphic, septate, and DES-related uteri.")
51
(b) the asymmetric obstructive form seen in three types:
unicornuate uterus with obstructed horn, double uterus with unilaterally obstructed horn, and double uterus with unilaterally obstructed vagina.

52
Class 4.—Unusual configurations and combinations of defects.
Imaging can display the morphology of unusual uterovaginal anomalies. An example is a case of didelphic uterus and longitudinal vaginal septum (lateral fusion defect) combined with obstructed hymen (vertical fusion defect)
 combined with obstructed hymen (vertical fusion defect)")
57
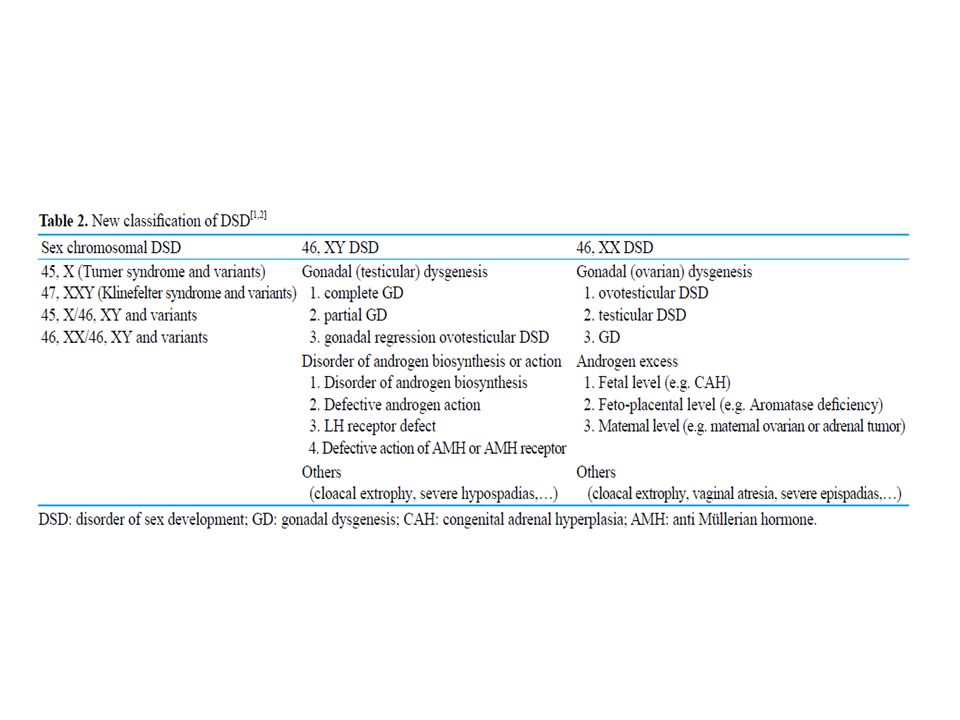
Disorders of sex development (DSD)
group of congenital conditions in which the development of the chromosomal, gonadal or anatomical sex has been atypical. DSD has replaced the formerly used term "intersex"

58
Ambiguous gender virilized female undervirilized male

59
Partial Androgen Insensitivity
Male

60
Congenital Adrenal Hyperplasia due to Steroid 21-Hydroxylase Deficiency
Three forms of CAH lead to ambiguity of the external genitalia in 46,XX patients: 21-hydroxylase deficiency, 11-hydroxylase deficiency and 3β-hydroxysteroid dehydrogenase 2 deficiency. Mutations in enzymes involved in adrenal steroid biosynthesis lead to glucocorticoid deficiency, with consequent increase in ACTH, resulting in adrenal androgen excess and adrenal hyperplasia. Female patients with CAH have intact female internal genitalia.

61
BOY ?????? GIRL

65
A. Disorders of gonadal (ovarian) development
1. Ovotesticular DSD, previously named true hermaphroditism. is a very rare disorder defined by the presence of both ovarian and testicular tissue in the same individual

67
2. Testicular DSD (SRY+, dup SOX9, RSPO1),
previously named XX males or XX sex reversal It is a rare syndrome affecting 1 in 20,000-25,000 newborn males.48 The majority of the cases are phenotypically normal males but in some cases there is genital ambiguity. Approximately 10-15% show various degrees of hypospadias.

68
Figure 1. Classes of hypospadias by location of the meatus
Figure 1. Classes of hypospadias by location of the meatus. (A) Anterior, on the inferior surface of the glans penis. (B) Coronal, in the balanopenile furrow. (C) Distal, on the distal third of the shaft. (D) Penoscrotal, at the base of the shaft in front of the scrotum. (E) Scrotal, on the scrotum or between the genital swellings. (F) Perineal, behind the scrotum or genital swellings.
 Anterior, on the inferior surface of the glans penis. (B) Coronal, in the balanopenile furrow. (C) Distal, on the distal third of the shaft. (D) Penoscrotal, at the base of the shaft in front of the scrotum. (E) Scrotal, on the scrotum or between the genital swellings. (F) Perineal, behind the scrotum or genital swellings.")
69
Female epispadia Epispadias is an uncommon and partial form of a spectrum of failures of abdominal and pelvic fusion in the first months of embryogenesis known as the exstrophy - epispadias complex

70
3. Gonadal dysgenesis In gonadal (ovarian) dysgenesis with normal XX karyotype, patients present with a female phenotype but fail to proceed to puberty and do not develop female secondary characteristics. They have elevated gonadotrophins and streak gonads.
 dysgenesis with normal XX karyotype, patients present with a female phenotype but fail to proceed to puberty and do not develop female secondary characteristics. They have elevated gonadotrophins and streak gonads.")
71
B. Androgen excess, previously named female
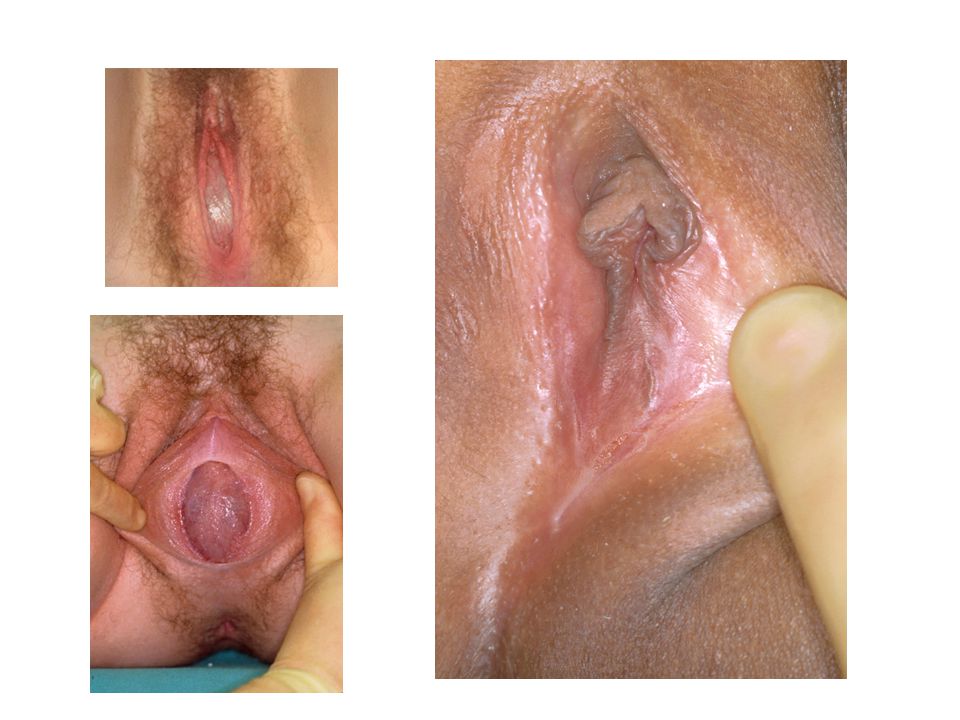
pseudohermaphroditism 1. Fetal (congenital adrenal hyperplasia, glucocorticoid receptor mutations) Affected girls may present with significant virilization of the external genitalia, indicating prenatal androgen excess.
 Affected girls may present with significant virilization of the external genitalia, indicating prenatal androgen excess.")
72
Prader Classification
of the Degree of Androgenization in a Female With CAH

74
Prader V, 14 y.o., sex identity -male, (non-salt losing form)
")
75
2. Fetoplacental (aromatase deficiency)
Aromatase deficiency (CYP19Α1: P450arom) Aromatase (P450arom) catalyses the conversion of androgens (C19 steroids) to estrogens (C18 steroids), 3. Maternal (luteoma, exogenous)
 Aromatase (P450arom) catalyses the conversion of androgens (C19 steroids) to estrogens (C18 steroids), 3. Maternal (luteoma, exogenous)")
76
C. Other abnormalities [cloacal exstrophy,
vaginal atresia, MURCS (Müllerian, renal, cervicothoracic somite abnormalities), other syndromes] This category includes anomalies such as vaginal atresia, cloacal exstrophy, uterine anomalies, (Müllerian agenesis/hypoplasia), labial adhesions.
, other. syndromes] This category includes anomalies such as vaginal. atresia, cloacal exstrophy, uterine anomalies, (Müllerian agenesis/hypoplasia), labial adhesions.")
77
Thank you

Similar presentations

















 become male or female?>")


















