Download presentation
Presentation is loading. Please wait.

1
Molecular Evolution Revised 29/12/06
Bioinformatics ICES 2006 Molecular Evolution Revised 29/12/06

2
Phylogeny is the inference of evolutionary relationships
All forms of life share a common origin. deduce the correct trees for all species of life to estimate the time of divergence between organisms since the time they last shared a common ancestor

3
Terminology Phylogenetic trees that are used to assess the relationships of homologous proteins (or nucleotide sequences) in a family Clade Bifurcating node Branch OUT or external node Internal node Phylogram

4
Terminology

5
Terminology Species tree versus gene tree
In a species tree an internal node represents a speciation event In a gene tree an internal node represents the divergence of an ancestral gene into two new genes with distinct sequences Species tree <> Gene tree horizontal gene transfer gene duplications

6
Species tree versus gene tree
Gray et al.

7
Phylogenetic inference
Selection of sequences for analysis Multiple sequence alignment Tree building Tree evaluation

8
Phylogenetic inference
selection of sequences for analysis DNA: Higher phylogenetic signal: Synonymous vs nonsynonymous substitutions (detect negative and positive selection) Protein: Phylogenetic signal less predominant than in DNA Better to construct a tree for evolutionary distant species or genes RNA: rRNA often used for constructing species trees
 Protein: Phylogenetic signal less predominant than in DNA. Better to construct a tree for evolutionary distant species or genes. RNA: rRNA often used for constructing species trees.")
9
Phylogenetic inference
2. multiple sequence alignment This is a critical step in the analysis as in many cases the alignment of amino acids or nucleotides in a column implies that they share a common ancestor If you misalign a group of sequences you will still be able to produce a tree. However, it is not likely to be biologically meaningful. Crap in is crap out! Inspect the alignment to be sure that all sequences are homologous Some times with ClustalW distantly related sequences are not well aligned. Try different gap and extension parameters to improve the alignment Only use these columns of the multiple alignment for which you have data for all organisms or sequences. Delete the columns for which this is not the case. Delete columns with gaps

10
Phylogenetic inference
3. Tree building Character-based methods Non-character based methods Methods based on an explicit model of evolution Maximum Likelihood Methods/Bayesian Phylogeny Pairwise distance methods Methods not based on an explicit Maximum Parsimony Methods

11
Distance based methods
calculate the distances between molecular sequences using some distance metric A clustering method (UPGMA, neighbour joining) is used to infer the tree from the pairwise distance matrix treat the sequence from a horizontal perspective, by calculating a single distance between entire sequences Advantage: Fast Allow using evolutionary models Disadvantage: sequences reduced to one number
 is used to infer the tree from the pairwise distance matrix. treat the sequence from a horizontal perspective, by calculating a single distance between entire sequences. Advantage: Fast. Allow using evolutionary models. Disadvantage: sequences reduced to one number.")
12
Character based methods
treat the sequences from a vertical perspective they search for each column of the alignment, the simplest explanation for how the characters evolved. For instance, MP involves a search for a tree with the fewest number of amino acid (or nucleotide character changes that account for the observed differences between the protein (gene) sequences.
 sequences.")
14
Phylogenetic inference
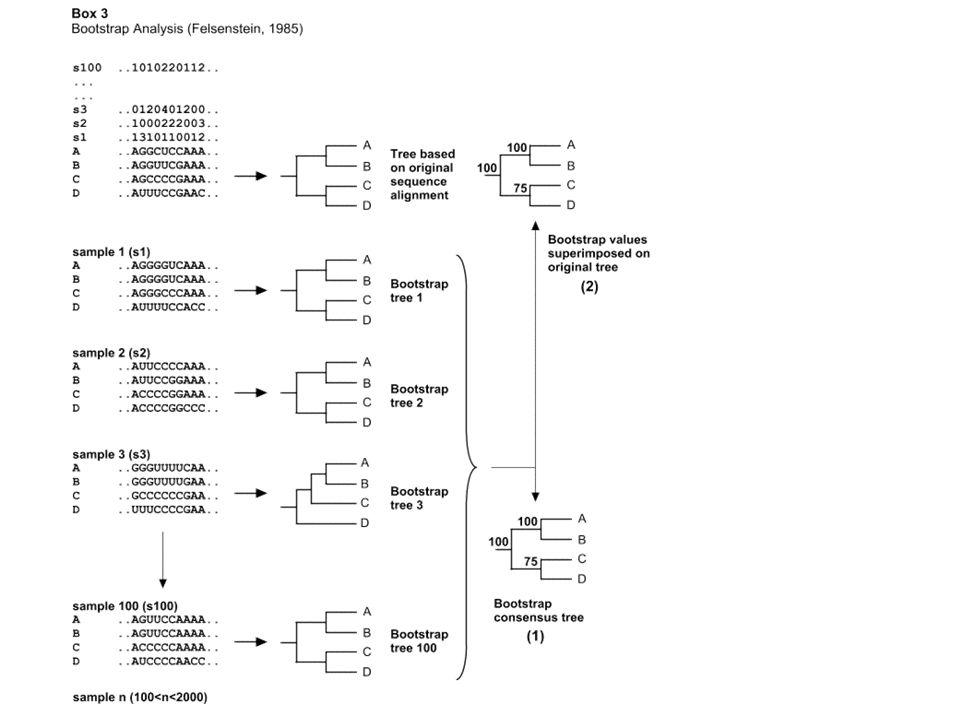
4. Tree evaluation: bootstrapping sampling technique for estimating the statistical error in situations where the underlying sampling distribution is unknown evaluating the reliability of the inferred tree - or better the reliability of specific branches How to proceed: From the original alignment, columns in the sequence alignment are chosen at random ‘sampling with replacement’ a new alignment is constructed with the same size as the original one a tree is constructed This process is repeated 100 of times

15
Phylogenetic inference
Show bootstrap values on phylogenetic trees majority-rule consensus tree map bootstrap values on the original tree

17
Maximum parsimony Principle
Select that tree that minimizes the total tree length = being the number of nucleic acid substitutions or amino acid replacements required to explain a given set of data. Method a particular topology is considered for this topology, the ancestral sequences at each branching point are reconstructed the minimum number of events to explain the sequence differences over the whole tree is computed: the minimum number of substitutions is computed for each nucleotide (or amino acid) site, and the numbers for all sites are added. another tree topology is chosen
 site, and the numbers for all sites are added. another tree topology is chosen.")
18
Maximum parsimony

19
Maximum parsimony OTU's rooted tree topologies
unrooted tree topologies 3 1 4 15 5 105 6 954 7 10395 8 135135 9 equation Exhaustive search impossible Heuristics needed

20
Maximum parsimony Find different tree topologies that are 'equally parsimonious‘ Represent results as a consensus tree. 'strict' consensus tree 'majority-rule' consensus tree

21
Maximum parsimony Only informative sites of the alignment are used in the construction of the tree: when there are at least two different kinds of characters, each represented at least two times

22
Maximum parsimony Parsimony trees are usually only represented as a tree topology (cladogram): sometimes, the parsimony program cannot decide in which branches the substitutions have been taken place. It can not calculate branch lengths.
: sometimes, the parsimony program cannot decide in which branches the substitutions have been taken place. It can not calculate branch lengths.")
23
Maximum parsimony Assumptions Equal rate of evolution in all branches
Advantages sequence information is not reduced to one number (such as for example in pairwise distance methods) Disadvantages of maximum parsimony methods can be slow for very large datasets no correction for multiple mutations, i.e. no substitution model can be applied (see further) sensitive to unequal rates of evolution in different lineages (see further) =>long branch attraction (voorbeeld hiervan?)
 Disadvantages of maximum parsimony methods. can be slow for very large datasets. no correction for multiple mutations, i.e. no substitution model can be applied (see further) sensitive to unequal rates of evolution in different lineages (see further) =>long branch attraction (voorbeeld hiervan )")
24
Pairwise distance methods
Distance calculation Inferring the tree topology

25
Pairwise distance methods
Distance calculation Approach: align pairs of sequences and count the number of differences (Hamming distance). For an alignment of length N with n sites at which there are differences: D= (n/N*100). Problem: observed differences <> actual genetic distances between the sequences. => dissimilarity is an underestimation of the true evolutionary distance, because of the fact that some of the sequence positions are the result of multiple events Solution: Use an evolutionary model that corrects for multiple mutations
. For an alignment of length N with n sites at which there are differences: D= (n/N*100). Problem: observed differences <> actual genetic distances between the sequences. => dissimilarity is an underestimation of the true evolutionary distance, because of the fact that some of the sequence positions are the result of multiple events. Solution: Use an evolutionary model that corrects for multiple mutations.")
26
Pairwise distance methods
Distance calculation

27
Pairwise distance methods
Distance calculation

28
Pairwise distance methods
Distance calculation Other evolutionary models

29
Pairwise distance methods
Tree inference: UPGMA Ultrametric trees are rooted trees, in which all the endnodes are equidistant from the root of the tree, Assuming a molecular clock: i.e, that all sequences evolve at a similar rate

30
Pairwise distance methods
Tree inference: UPGMA when two OTUs are grouped, we treat them as a new single OTU when OTUs A, B (which have been grouped before) and C are grouped into a new node ‘u’, then the distance from node ‘u’ to any other node ‘k’ (e.g. grouping D and E) is simply computed as follows:
 and C are grouped into a new node ‘u’, then the distance from node ‘u’ to any other node ‘k’ (e.g. grouping D and E) is simply computed as follows:")
31
Pairwise distance methods
Tree inference: UPGMA

32
Pairwise distance methods
Tree inference: UPGMA Advantages: Fast Allows incorporation of evolutionary models Disadvantages: Assumption of a molecular clock

33
Pairwise distance methods
Tree inference: neighbor joining Additive distances can be fitted to an unrooted tree such that the evolutionary distance between a pair of OTUs equals the sum of the lengths of the branches connecting them, rather than being an average as in the case of cluster analysis Tree construction methods: minimum evolution, the tree that minimizes the sum of the lengths of the branches is regarded the best estimate of the phylogeny Drawback for the ME method: is that in principle all different tree topologies have to be investigated in order to find the ‘minimum’ tree. The neighbour joining (NJ) method, developed by Saitou and Nei (1987) offers a heuristic approach to solve this problem
 method, developed by Saitou and Nei (1987) offers a heuristic approach to solve this problem.")
34
Pairwise distance methods
Tree inference: neighbor joining

35
Pairwise distance methods
Tree inference: neighbor joining

36
Pairwise distance methods
Tree inference: neighbor joining

37
Pairwise distance methods
Tree inference: neighbor joining Advantages: Fast Allows incorporation of evolutionary models No assumption of a molecular clock

Similar presentations

 or Evolutionary tree represents the evolutionary relationships among a set of organisms or groups of.>")











 relieves that restriction, but still enforces.>")

 and likelihood methods, pairwise distance methods form the third large group of methods to infer evolutionary trees.>")




