Download presentation
Presentation is loading. Please wait.

2
Aim in building a phylogenetic tree is to use a knowledge of the characters of organisms to build a tree that reflects the relationships between them. Organisms with many characters in common are more likely to be related than those with few in common.

3
We want to use characters that are homologous [shared because of common ancestry] rather than analagous [independently evolved]. But how is this to be done? Turns out that there are many approaches the first of which is to apply parsimony.
![ We want to use characters that are homologous [shared because of common ancestry] rather than analagous [independently evolved].](http://images.slideplayer.com/15/4549841/slides/slide_3.jpg " But how is this to be done. Turns out that there are many approaches the first of which is to apply parsimony..")
4
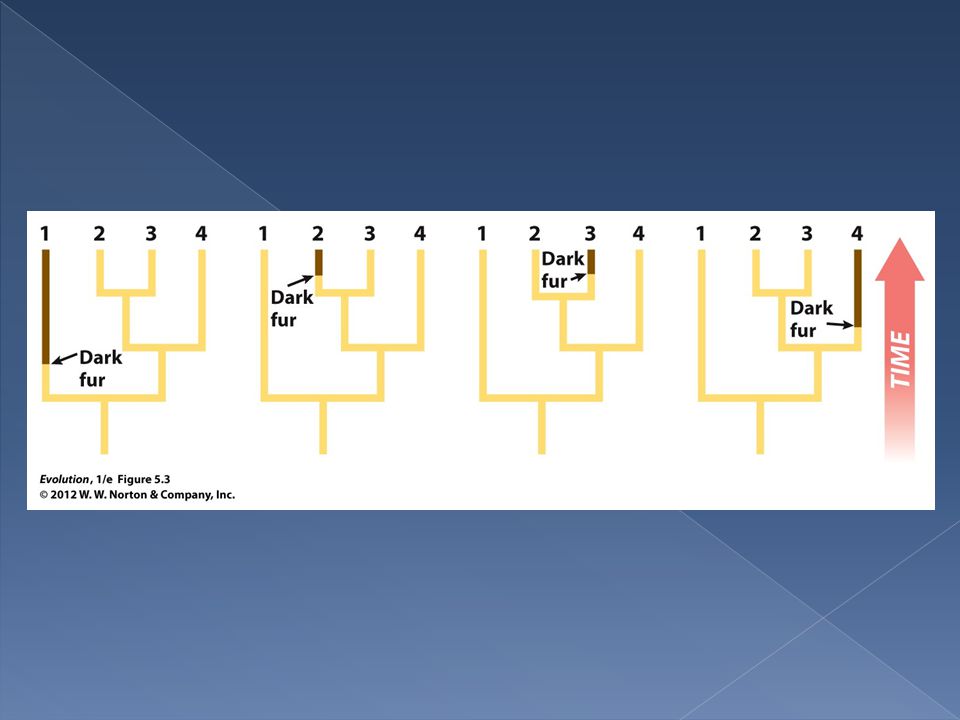
The basic idea of parsimony in tree building is to build a tree that requires the fewest evolutionary changes in its construction. In the following trees one species differs from the other three. In each tree a single evolutionary change is all that is required to build it.

6
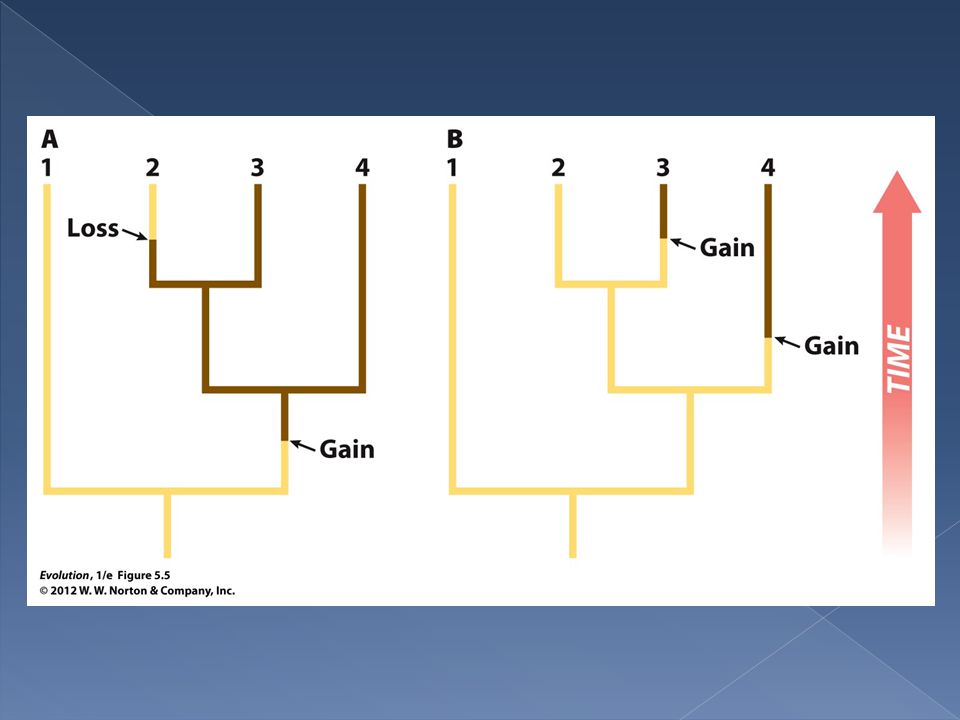
Similarly, we can [in the next slide] analyze a situation where two non-sister taxa (3&4) share a trait. There are two equally likely explanations in this case.
![ Similarly, we can [in the next slide] analyze a situation where two non-sister taxa (3&4) share a trait.](http://images.slideplayer.com/15/4549841/slides/slide_6.jpg " There are two equally likely explanations in this case..")
8
The same logic applies when dealing with multiple traits (3 traits each with two states in the next example).
.")
10
Each trait is treated separately and the most parsimonious explanation is calculated.

12
When the data are pooled a total of five changes are present on the tree.

14
Its turns out that the tree we just dealt with is not the most parsimonious tree. It is possible to build a tree that has only three changes [it is impossible to have fewer than three changes].

16
In the previous example it was easy to see the minimum number of changes needed to make a most parsimonious tree. For larger trees this is not so simple to do. The Fitch algorithm can be used to figure the minimum number of changes necessary for a given tree.

17
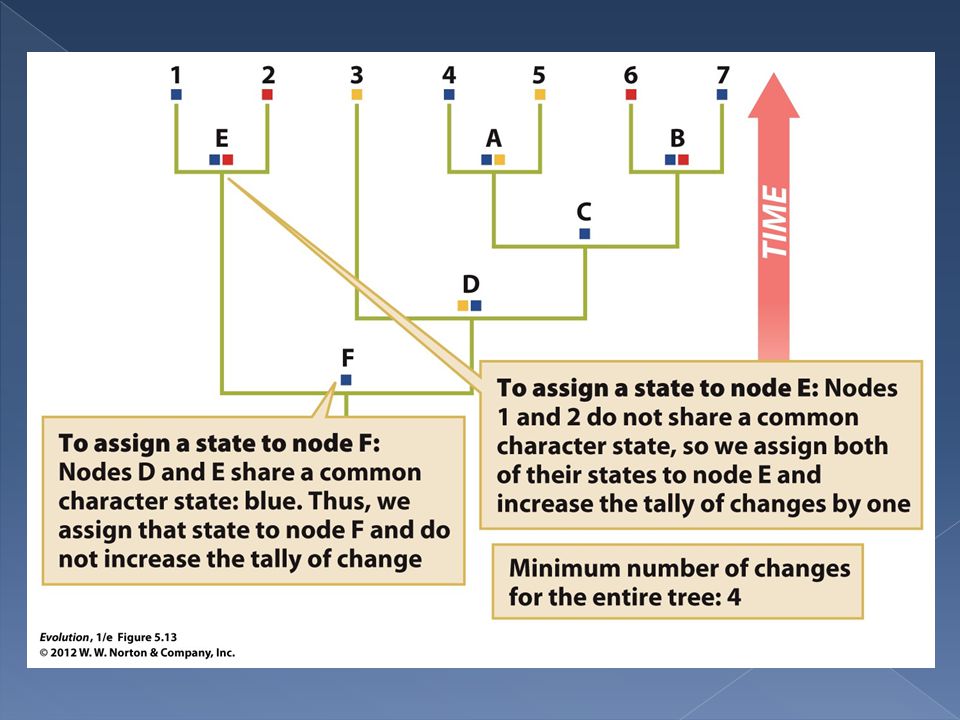
The Fitch algorithm begins at the branch tips of a tree and proceeds towards the base of the tree. A running count is kept of the number of the character changes needed.

19
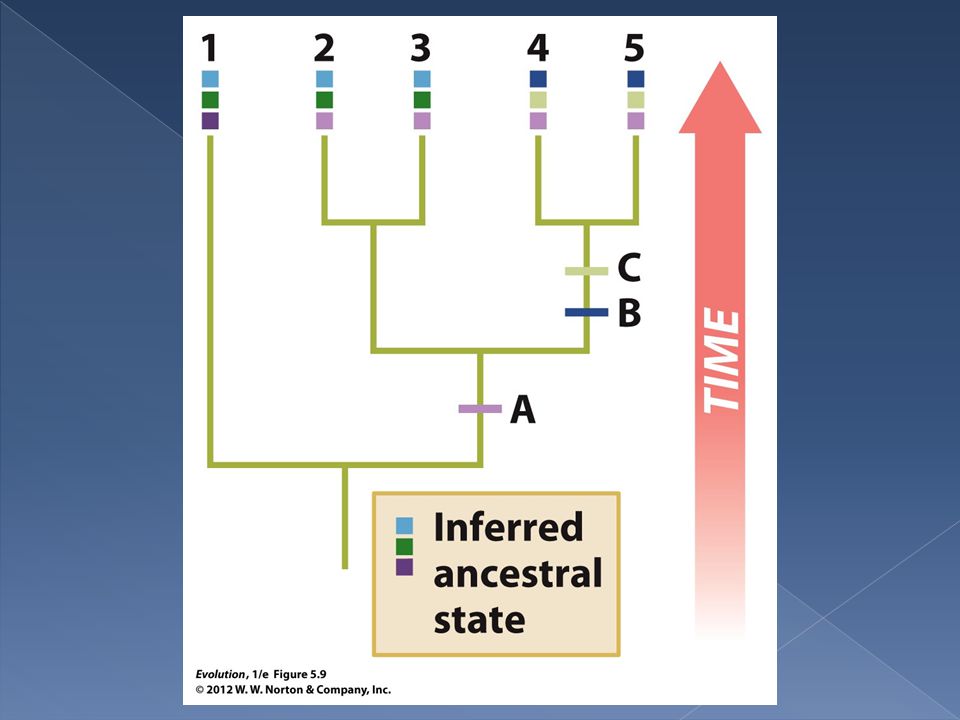
As we proceed down the tree each internal node is assigned one or more character states. Two rules are used to assign character states at nodes.

20
Rule 1. If the two daughters of a node share no stated in common we assign to the node all possible states for both daughters. In other words the set of possible traits at the node is the union of the sets of possible traits for daughters 1 and 2. In this case we increase the tally of character changes by one.

22
Rule 2. If the daughters of a node share one or more possible states of a trait then we assign the shared states to the node. In other words we assign the intersection of the sets of possible states for each daughter to the node. In this case we do not increase the tally of character changes.

25
The Fitch algorithm just tells us the minimum number of changes needed for a given tree. It does not tell us if a different tree would have fewer. In order to compare different trees to find the most parsimonious we would have to repeat the Fitch process for all the trees.

26
Another approach to building phylogenetic trees is to use distance methods. In this approach pairwise distances, (where distance is a measure of morphological or genetic differences between species) are calculated and used in tree construction.
 are calculated and used in tree construction..")
27
Distances can be: › Counts of the number of character differences between species. › Based on morphological measurements › In living species most commonly counts of base pair differences in DNA sequences or amino acid differences coded for are used to build trees.

29
Because insertion/deletion mutations occur and can shift the reading frame of a length of DNA sometimes sequences need to be aligned before using them to build a phylogenetic tree.

31
Once distance measures have been calculated the pairwise measures (differences between individual pairs of species) are arranged into a distance matrix.
 are arranged into a distance matrix.")
33
Once distance measures are tabulated we need to figure out how to arrange these data on a tree and decide how long to make the branches. For four species there is only one basic tree shape and only three pairwise species arrangements.

35
There are multiple statistical procedures that can be used to construct trees using distance data. The details of these are beyond the scope of this class. However, the aim of all of them is to find a tree topology (or structure) in which each pairwise distance in the tree is as close as possible to that in the data matrix.
 in which each pairwise distance in the tree is as close as possible to that in the data matrix..")
38
One philosophical objection to trees built using distance methods is that they don’t explicitly incorporate underlying evolutionary relationships. They are similarity measures (and assume that similarity reflects homology), but analagous traits may sometimes be used.
, but analagous traits may sometimes be used..")
39
We have spent a lot of time looking at ways of assessing how well trees are supported by data. However, the big challenge in building phylogenies is in identifying potentially useful trees from the huge number of potential trees

40
It turns out that the number of potential phylogenetic trees increases exponentially with the number of taxa in the tree.

42
The challenge for phylogenticists who cannot search every possible tree is to develop strategies to search only for plausible trees. Very computer intensive algorithms are used to do this, but the underlying methodologies are beyond the scope of this class.

43
Phylogenetic trees are hypotheses about the relationships between taxa. Once a tree is constructed how much confidence can we have that the tree (or some part of it) is correct? This is an issue of statistical confidence.
 is correct. This is an issue of statistical confidence..")
44
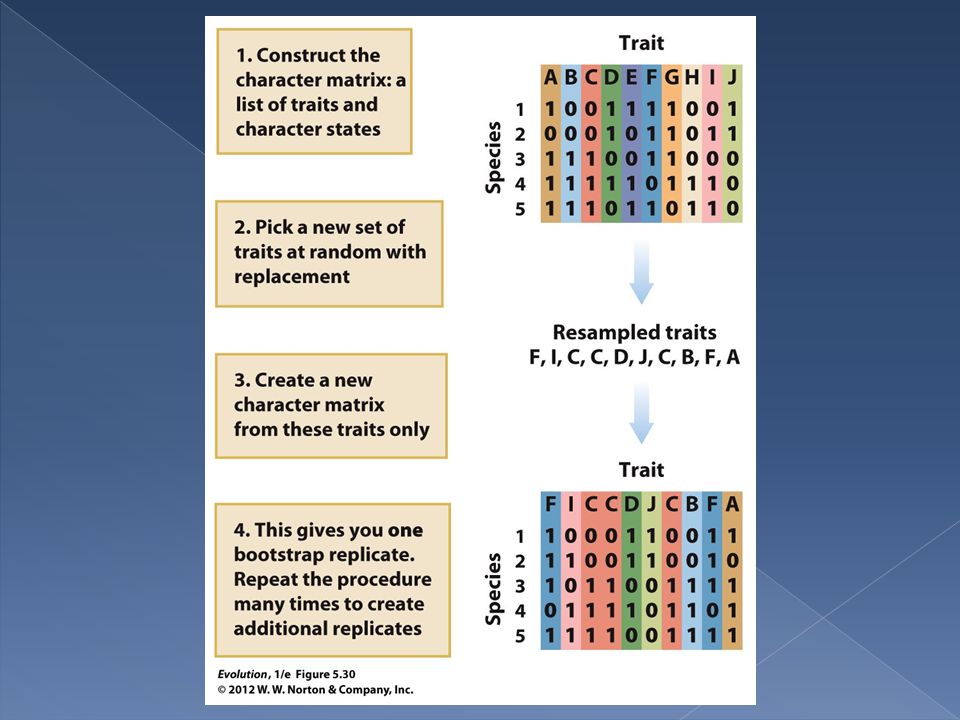
There are a number of techniques that scientists have developed to measure how well the data support a given tree. One of the most widely used is bootstrap resampling.

45
Bootstrap resampling is based on the idea that the data set that the phylogeny is based on is itself only one possible set of data that the tree could have been built with. How sensitive is the tree’s structure to the set of data we used? If we had used a similar but not identical set of data would we have produced the same tree?

46
To carry out a bootstrap analysis we simply resample from our original character matrix. We randomly pick sets of traits with replacement from our data set and the new data matrix is used to build a phylogenetic tree. That tree is then compared to the original tree.

49
After repeated bootstrap resamplings we see how often the new trees match the original tree. If resampled trees match the original tree 90% of the time we say the tree has 90% bootstrap support.

51
For a considerable period of time before widespread genomic analysis there was controversy about whether the closest relatives of the eutherian (or placental) mammals were the marsupials or the monotremes.
 mammals were the marsupials or the monotremes.")
53
In 2001 Killian et al. sequenced a large nuclear gene from 11 species of placental mammal, two marsupials and two monotremes. Using the sequence data they constructed a phylogeny of the mammals that indicated the placental and marsupial mammals were sister groups.

55
To check how strongly their data supported the monophyly of the placental and marsupial mammals Killian et al. carried out a bootstrap resampling analysis of their data. The results showed that the marsupials and placental mammals formed a monophyletic clade in 100% of the trees.

56
The bootstrap analysis thus indicated that strongly supported for this data set the monophyly of the placental and marsupial mammals. Since Killian’s paper numerous other studies of nuclear DNA have supported this conclusion.

Similar presentations


















 or Evolutionary tree represents the evolutionary relationships among a set of organisms or groups of.>")









 relieves that restriction, but still enforces.>")
 traits are useful in understanding evolutionary relationships Shared primitive (plesiomorphic)>")
 Phylogenetic Trees (I) Maximum Parsimony.>")

: A C A A C C A A C A C C 2 0 1 0 0 Sum of branch lengths = total number of changes.>")
 Arrive promptly at lab - need to leave quickly.>")

.>")

