Download presentation
Presentation is loading. Please wait.

2
Thalassemia Dr.Alireza Nikanfar Hematology and oncology research center of Tabriz University of Medical Sciences

3
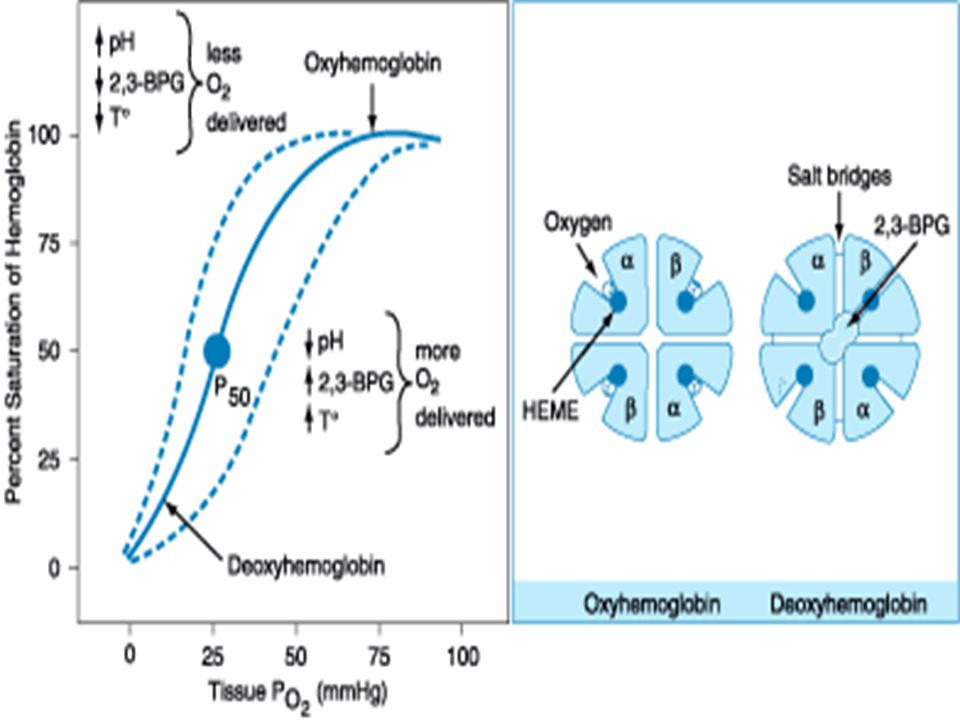
Hemoglobin Structure tetramer of globin polypeptide chains: a pair of α -like chains 141 amino acids long and a pair of ß -like chains 146 amino acids long tetramer of globin polypeptide chains: a pair of α -like chains 141 amino acids long and a pair of ß -like chains 146 amino acids long The major adult hemoglobin, HbA, has the structure α 2 ß 2. HbF (α 2 γ 2 ) predominates during most of gestation, and HbA2 (α 2 δ 2 ) is a minor adult hemoglobin. The major adult hemoglobin, HbA, has the structure α 2 ß 2. HbF (α 2 γ 2 ) predominates during most of gestation, and HbA2 (α 2 δ 2 ) is a minor adult hemoglobin.
 predominates during most of gestation, and HbA2 (α 2 δ 2 ) is a minor adult hemoglobin. The major adult hemoglobin, HbA, has the structure α 2 ß 2. HbF (α 2 γ 2 ) predominates during most of gestation, and HbA2 (α 2 δ 2 ) is a minor adult hemoglobin..")
4
Hemoglobin Structure Each globin chain enfolds a single heme moiety, consisting of a protoporphyrin IX ring complexed with a single iron atom in the ferrous state (Fe 2 +), Each globin chain enfolds a single heme moiety, consisting of a protoporphyrin IX ring complexed with a single iron atom in the ferrous state (Fe 2 +), Each heme moiety can bind a single oxygen molecule; every molecule of hemoglobin can thus transport up to four oxygen molecules. Each heme moiety can bind a single oxygen molecule; every molecule of hemoglobin can thus transport up to four oxygen molecules.

8
Thalassemia Syndromes inherited disorders of α- or ß -globin biosynthesis inherited disorders of α- or ß -globin biosynthesis The reduced supply of globin diminishes production of hemoglobin tetramers, causing hypochromia and microcytosis. The reduced supply of globin diminishes production of hemoglobin tetramers, causing hypochromia and microcytosis. Unbalanced chain accumulation dominates the clinical phenotype Unbalanced chain accumulation dominates the clinical phenotype

9
α-Thalassemia Syndromes α-Thalassemia Syndromes α-thalassemia-2 trait, in which one of the four α- globin loci is deleted α-thalassemia-2 trait, in which one of the four α- globin loci is deleted α -thalassemia-1 trait, with two deleted loci α -thalassemia-1 trait, with two deleted loci HbH disease, with three loci deleted HbH disease, with three loci deleted hydrops fetalis with Hb Bart's, with all four loci deleted hydrops fetalis with Hb Bart's, with all four loci deleted Nondeletion forms Nondeletion forms

11
α-Thalassemia Syndromes α -Thalassemia-2 trait is an asymptomatic, silent carrier state. α -Thalassemia-2 trait is an asymptomatic, silent carrier state. α -Thalassemia-1 trait resembles -thalassemia minor α -Thalassemia-1 trait resembles -thalassemia minor Heterozygosity for a deletion that removes both genes from the same chromosome (cis deletion) is common in Asians and Mediterranean individuals, as is homozygosity for -thalassemia-2 (trans deletion). Both produce asymptomatic hypochromia and microcytosis. Heterozygosity for a deletion that removes both genes from the same chromosome (cis deletion) is common in Asians and Mediterranean individuals, as is homozygosity for -thalassemia-2 (trans deletion). Both produce asymptomatic hypochromia and microcytosis.
 is common in Asians and Mediterranean individuals, as is homozygosity for -thalassemia-2 (trans deletion). Both produce asymptomatic hypochromia and microcytosis. Heterozygosity for a deletion that removes both genes from the same chromosome (cis deletion) is common in Asians and Mediterranean individuals, as is homozygosity for -thalassemia-2 (trans deletion). Both produce asymptomatic hypochromia and microcytosis..")
13
α-Thalassemia Syndromes In HbH disease, HbA production is only 25 to 30% of normal In HbH disease, HbA production is only 25 to 30% of normal In adults, unpaired chains accumulate and are soluble enough to form ß 4 tetramers called HbH In adults, unpaired chains accumulate and are soluble enough to form ß 4 tetramers called HbH Patients with HbH disease have thalassemia intermedia characterized by moderately severe hemolytic anemia but milder ineffective erythropoiesis Patients with HbH disease have thalassemia intermedia characterized by moderately severe hemolytic anemia but milder ineffective erythropoiesis Survival into midadult life without transfusions is common. Survival into midadult life without transfusions is common.

18
α-Thalassemia Syndromes The homozygous state for the α-thalassemia-1 cis deletion (hydrops fetalis) causes total absence of α-globin synthesis The homozygous state for the α-thalassemia-1 cis deletion (hydrops fetalis) causes total absence of α-globin synthesis Excess γ globin forms tetramers called Hb Bart's (γ 4 ), which has an extraordinarily high oxygen affinity Excess γ globin forms tetramers called Hb Bart's (γ 4 ), which has an extraordinarily high oxygen affinity It delivers almost no O 2 to fetal tissues, causing tissue asphyxia, edema (hydrops fetalis), congestive heart failure, and death in utero. It delivers almost no O 2 to fetal tissues, causing tissue asphyxia, edema (hydrops fetalis), congestive heart failure, and death in utero.
, congestive heart failure, and death in utero..")
19
ß -Thalassemia Syndromes Mutations causing thalassemia can affect any step in the pathway of globin gene expression: Mutations causing thalassemia can affect any step in the pathway of globin gene expression: Hypochromia and microcytosis Hypochromia and microcytosis In heterozygotes ( ß -thalassemia trait), this is the only abnormality seen; anemia is minimal. In heterozygotes ( ß -thalassemia trait), this is the only abnormality seen; anemia is minimal.
, this is the only abnormality seen; anemia is minimal..")
22
ß -Thalassemia Syndromes In homozygous states accumulation of highly insoluble unpaired α chains, which form toxic inclusion bodies that kill developing erythroblasts in the marrow ineffective erythropoiesis In homozygous states accumulation of highly insoluble unpaired α chains, which form toxic inclusion bodies that kill developing erythroblasts in the marrow ineffective erythropoiesis The few surviving red cells bear a burden of inclusion bodies, detected in the spleen, shortening the red cell life span and producing severe hemolytic anemia. The few surviving red cells bear a burden of inclusion bodies, detected in the spleen, shortening the red cell life span and producing severe hemolytic anemia. Erythroid hyperplasia can become exuberant and produce extramedullary erythropoietic tissue in the liver and spleen Erythroid hyperplasia can become exuberant and produce extramedullary erythropoietic tissue in the liver and spleen

23
ß -Thalassemia Syndromes Massive bone marrow expansion deranges growth and development. Massive bone marrow expansion deranges growth and development. "chipmunk" facies "chipmunk" facies thinning and pathologic fracture of long bones and vertebrae due to cortical invasion by erythroid elements, thinning and pathologic fracture of long bones and vertebrae due to cortical invasion by erythroid elements, profound growth retardation profound growth retardation

24
ß -Thalassemia Syndromes Hemolytic anemia causes hepatosplenomegaly, leg ulcers, gallstones, and high-output congestive heart failure. Hemolytic anemia causes hepatosplenomegaly, leg ulcers, gallstones, and high-output congestive heart failure. The conscription of caloric resources to support erythropoiesis leads to inanition, susceptibility to infection, endocrine dysfunction, and, in the most severe cases, death during the first decade of life. The conscription of caloric resources to support erythropoiesis leads to inanition, susceptibility to infection, endocrine dysfunction, and, in the most severe cases, death during the first decade of life.

25
ß -Thalassemia Syndromes Severity is highly variable Severity is highly variable Alleles associated with milder synthetic defects and coinheritance of Alleles associated with milder synthetic defects and coinheritance of α-thalassemia trait reduce clinical severity HbF persists to various degrees in thalassemias. HbF persists to various degrees in thalassemias.

27
ß -Thalassemia Syndromes ß -thalassemia major ß -thalassemia major ß -thalassemia intermedia can survive without transfusion ß -thalassemia intermedia can survive without transfusion ß -thalassemia minor and ß -thalassemia trait describe asymptomatic heterozygotes for ß thalassemia ß -thalassemia minor and ß -thalassemia trait describe asymptomatic heterozygotes for ß thalassemia

31
Diagnosis The diagnosis of ß -thalassemia major is readily made during childhood on the basis of severe anemia accompanied by hepatosplenomegaly; profound microcytosis; a characteristic blood smear ; and elevated levels of HbF, HbA2, or both. The diagnosis of ß -thalassemia major is readily made during childhood on the basis of severe anemia accompanied by hepatosplenomegaly; profound microcytosis; a characteristic blood smear ; and elevated levels of HbF, HbA2, or both. Patients with ß -thalassemia intermedia exhibit similar stigmata but can survive without chronic hypertransfusion. Patients with ß -thalassemia intermedia exhibit similar stigmata but can survive without chronic hypertransfusion.

33
Diagnosis ß -Thalassemia minor (i.e., ß thalassemia trait) usually presents as profound microcytosis and hypochromia with target cells but only minimal or mild anemia ß -Thalassemia minor (i.e., ß thalassemia trait) usually presents as profound microcytosis and hypochromia with target cells but only minimal or mild anemia The mean corpuscular volume is rarely >75 fL; the hematocrit is rarely 75 fL; the hematocrit is rarely <30 to 33%. Hemoglobin electrophoresis classically reveals an elevated HbA2 (3.5 to 7.5%), but some forms are associated with normal HbA2 and/or elevated HbF. Hemoglobin electrophoresis classically reveals an elevated HbA2 (3.5 to 7.5%), but some forms are associated with normal HbA2 and/or elevated HbF.
, but some forms are associated with normal HbA2 and/or elevated HbF. Hemoglobin electrophoresis classically reveals an elevated HbA2 (3.5 to 7.5%), but some forms are associated with normal HbA2 and/or elevated HbF..")
34
Diagnosis Persons with α-thalassemia trait may exhibit mild hypochromia and microcytosis, usually without anemia Persons with α-thalassemia trait may exhibit mild hypochromia and microcytosis, usually without anemia HbA2 and HbF levels are normal. HbA2 and HbF levels are normal. HbH disease resembles ß -thalassemia intermedia, with the added complication that the HbH molecule behaves like a moderately unstable hemoglobin. HbH disease resembles ß -thalassemia intermedia, with the added complication that the HbH molecule behaves like a moderately unstable hemoglobin.

39
خسته نباشید متشکرم

Similar presentations
























 Hb is found in RBCs its main function is to transport O2 to tissues. Structure: 2 parts : heme + globin Globin: four globin chains (2 α.>")



 is composed of four protein chains, two α and two β globin chains arranged into.>")


; FRCPEdin; FRCSEdin>")




 Hb is found in RBCs its main function is to transport O2 to tissues. Structure: 2 parts : heme + globin Globin: four chains. Heme: porphyrin.>")


 Regionβ-Thalassaemiaα 0 -Thalassaemiaα + -Thalassaemia Americas 0–30–50–40.>")