Download presentation
Presentation is loading. Please wait.

1
Prion Diseases

19
Kuru

21
Kuru figures Incidence 1 % (population aprox. 15,000) total F : M = 10 : 1 < 20 years of age F : M = 3 : 1 (20 % of cases) 20-40 years of age F : M = 15 : 1
 total F : M = 10 : 1 < 20 years of age F : M = 3 : 1 (20 % of cases) years of age F : M = 15 : 1.")
22
Etiology of Kuru Primary infectious: no fever, no CSF cell raise Genetical disease: –Pro: in families, in patients who moved out of Fore region. –Con: also in patients who came to live in Fore region, as well in young as in old patients. Toxic/deficiency: no toxic compound isolated, balanced diet. Endocanibalism

27
Sporadic CJD 1920/1921

33
Sporadic Creutzfeldt-Jakob disease diagnosis Rare disease: 1/10 6 Duration: short (months) Neurological signs and symptoms: Rapidly progressive dementia, myoclonus, ataxia, central visual disturbances, extrapyramidal signs, pyramidal signs, akinetic mutism (variant: chorea, dysesthesias, psychiatric disturbances) EEG and MRI Neuropathology WHO criteria: –Type of CJD (sporadic, genetic, iatrogenic, variant) –Strength of diagnosis (definite, probable, possible)
 Neurological signs and symptoms: Rapidly progressive dementia, myoclonus, ataxia, central visual disturbances, extrapyramidal signs, pyramidal signs, akinetic mutism (variant: chorea, dysesthesias, psychiatric disturbances) EEG and MRI Neuropathology WHO criteria: –Type of CJD (sporadic, genetic, iatrogenic, variant) –Strength of diagnosis (definite, probable, possible)")
34
MRI Proton density Weighted T2 EEG abnormalities normalsCJD

35
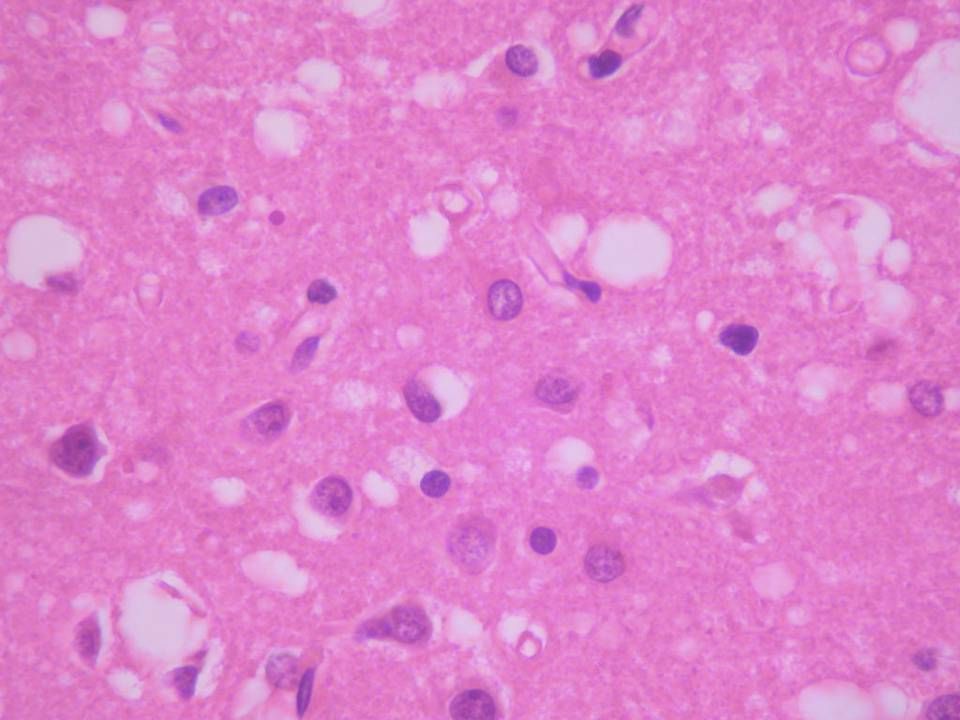
Gross sCJD

36
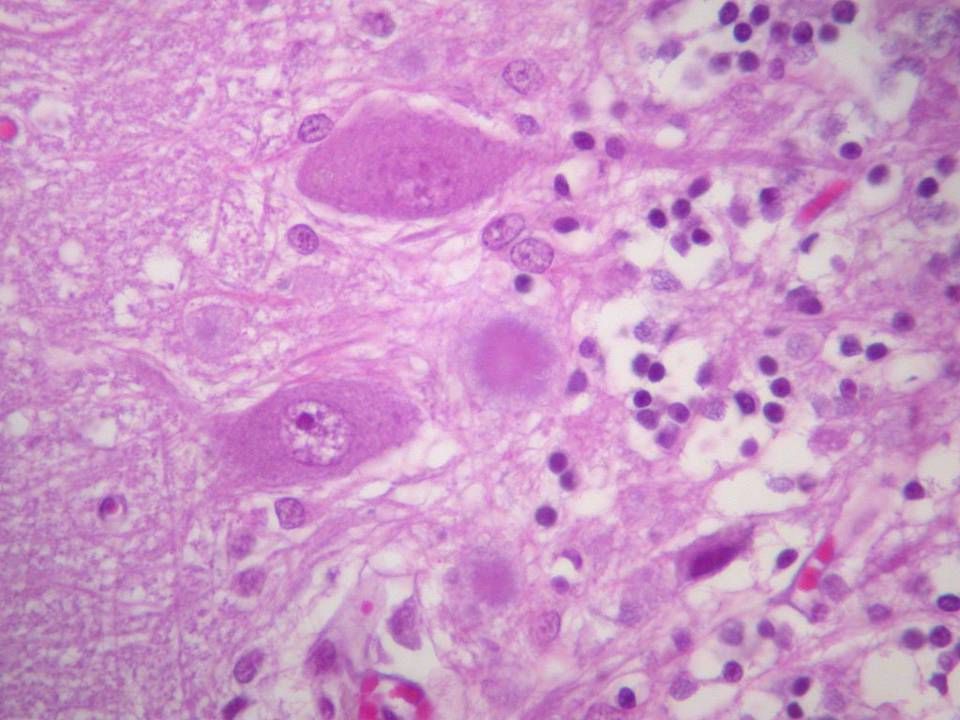
Micro sCJD

37
Prion Diseases/Transmissible Spongiform Encephalopathies CWD TME BSE FSE ex-TSE TSE's in animals sCJD fCJD GSS FFI Kuru iCJD vCJD TSE's in man Prion diseases 85% } 14% } 1% sporadic genetic acq. by infection ex-Scr Scrapie

38
Etiology Possibilities (ca 1980): Degenerative/HereditaryDegenerative/Hereditary (slow)Virus (slow)Virus
: Degenerative/HereditaryDegenerative/Hereditary (slow)Virus (slow)Virus")
39
Againts virus hypothesis Very resistant agent: Very resistant agent: resists dry heat ( > 200ºC) resists dry heat ( > 200ºC) resists steam autoclaving (up to134ºC, 18 mins.) resists steam autoclaving (up to134ºC, 18 mins.) resists formaldehyde resists formaldehyde resists UV-radiation resists UV-radiation resists Gamma-radiation ( > 0.3 MGray) resists Gamma-radiation ( > 0.3 MGray) etc. etc.

40
Prion docterine (Prusiner) In biochemical separation-infection studies: Infectivity is not present in a DNA/RNA fraction Infectivity is present in a protein fraction In conclusion: A protein (and that protein only) causes a prion disease
 In biochemical separation-infection studies: Infectivity is not present in a DNA/RNA fraction Infectivity is present in a protein fraction In conclusion: A protein (and that protein only) causes a prion disease")
41
From Prion gene to Prion protein 129 219 -8 E/K M/V 91 51 221 231254 Glycin - prolin rich sequency sign. peptide PRNP PrP c /PrP Sc (33 - 35 kD) S GPI SS CHO S GPI SS CHO codon PrP Sc (27- 30 kD) Proteinase K PrP c -fragm. hydrophob. seq. polymorph. 5 OR α2α2 α3α3 α1α1ß1ß1ß2ß2 α2α2 α3α3 α1α1ß1ß1ß2ß2 α2α2 α3α3 α1α1ß1ß1ß2ß2
 S GPI SS CHO S GPI SS CHO codon PrP Sc ( kD) Proteinase K PrP c -fragm. hydrophob. seq. polymorph. 5 OR α2α2 α3α3 α1α1ß1ß1ß2ß2 α2α2 α3α3 α1α1ß1ß1ß2ß2 α2α2 α3α3 α1α1ß1ß1ß2ß2.")
42
PrP C 129 Sh1 H1 106-126 Sh2 H2 H3 Octa-repeat GPI N-terminus

43
Abnormal folding of a protein protein (and that protein only) causes a prion disease PrP C to PrP Sc
 causes a prion disease PrP C to PrP Sc")
44
Conversion models for PrP C to PrP Sc Polymerisation PrP C PrP* PrP Sc PrP* Dimerisation PrP Sc PrP C PrP* PrP Sc PrP* PrP Sc …and subsequent breaking and seeding.

45
Structural assembly of PrP Sc

46
Aggregation

48
Crystal /Scrapie associated fibril

50
IHC for PrP in sCJD-neocortex

52
The “Z” family IV III II I Clin: dementia, ataxia, hypokinesia

53
Example of gCJD in cerebellum Dominant inheritance with dementia and ataxia mostly

54
Familial CJD vs Familial Fatal Insomnia 178 Asn- 129 V-f-CJD 178 Asn- 129 M-FFI

55
Prion diseases acquired through infection Kuru iatrogenic CJD new variant CJD

56
iCJD numberincubation time clinical presentation Intracerebral cornea neurosurgery stereotactic EEG Cerebral surface dura mater* Peripheral blood growth hormone gonadotrophin 4 5 2 174 180 4 17 mo 18 mo 6-12 y 12 y 13 y dementia/ataxia ataxia * Mostly Lyodura, now 2 cases with 0.1 N NaOH treated Tutoplast

57
Bovine Spongiform Encephalopathy

58
BSE medulla oblongata

59
BSE-microscopy

60
Clinical history in v-CJD 65% of cases onset with ‘psychiatric’ complaints: Behavioural disturbance Attention deficit Personality changes Depression Later weeks - months: Dysaesthesia (20 % onset with dysaesth) Ataxia Myoclonus/choreo-athetosis Progressive dementia No typical EEG 14-3-3 protein 50% of the cases positive bilateral Pulvinar sign on MRI
 Ataxia Myoclonus/choreo-athetosis Progressive dementia No typical EEG protein 50% of the cases positive bilateral Pulvinar sign on MRI")
61
MRI Flair sCJDFlair vCJD

62
Age Comparison with sp-CJD

63
vCJD autopsy

64
Tonsil biopsies in vCJD

65
Variant CJD

66
vCJD caused by BSE?

67
Strain typing Glycoform pattern Genotype/polymorphisms Clinical profile Inoculation in experimental animals: –Incubation time –Pathology profile

68
CH sidechains of PrP

69
Glycoform analysis sCJD vCJD/BSE

70
Survival after inoculation

71
Lesion profiling example

72
Lesion profiling Scrapie BSE FSE, exTSEvCJD spCJD

73
What’s the problem with Prion diseases Infectious material (whatever the origin: sporadic, genetic or infectious derived) Poorly disinfectable: –formic acid 96% 1 hr –2 N NaOH 1 hr –20,000 ppm Chlorine 1 hr
 Poorly disinfectable: –formic acid 96% 1 hr –2 N NaOH 1 hr –20,000 ppm Chlorine 1 hr")
74
Smallest infectious dose Smallest infectious dose = 300-600 kD = 14-28 PrP Sc molec. ID 50 /kD kD

76
Pathogenesis of infection 1.Direct brain contact 2.Vascular inoculation 3.Oral (intestinal) inoculation
 inoculation")
77
Role for autonomic nervous system 1997: infectivity after oral challenge in hamster first seen at T7 level of spinal cord (Diringer) 2002: PrP Sc seen in vagus nerve and splanchnic nerve before reaching spinal cord after oral challenge (Beekes)
 2002: PrP Sc seen in vagus nerve and splanchnic nerve before reaching spinal cord after oral challenge (Beekes)")
78
Role for hematologic system 1996: immunodeficient mice develop no prion disease after peripheral inoculation (Bruce)-spleen dependency 1991: FDCs found positive for PrP Sc in mice (Kitamoto) 1997: crucial role for B-cells in CJD pathogenesis (Aguzzi) 1998: PrP expression in B-cells not necessarry for CJD pathogenesis (Aguzzi) –exit B-cells
-spleen dependency 1991: FDCs found positive for PrP Sc in mice (Kitamoto) 1997: crucial role for B-cells in CJD pathogenesis (Aguzzi) 1998: PrP expression in B-cells not necessarry for CJD pathogenesis (Aguzzi) –exit B-cells")
79
Proposed route of peripheral infection M-cell Macroph. FDC Macroph. FDC X th CN Symp NS Brainstem Spinal cord Spleen lymphatics Brain > Gut GALT B-cell ++

80
PrP Sc pathogenesis: loss or toxic gain of function? PrP properties: Proposed function: ligand Cu 2+ binding (metallo-protein) Hydrophobic section: PrP cmt
 Hydrophobic section: PrP cmt.")
81
PrP protein-copper binding sites Manlgcwmlvlfvatwsdlglckkrpkpggwnt ggsrypgqgspggnryppqggggwgqphgggwg qphgggwgqphgggwgqphgggwgqgggthsqw nkpskpktnmkhmagaaaagavvgglggymlgs amsrpiihfgsdyedryyrenmhrypnqvyyrp mdeysnqnnfvhdcvnitikqhtvttttkgenf tetdvkmmervveqmcitqyeresqayyqrgss mvlfssppvillisfliflivg Cu binding motiv: hgggw or ggth

82
Octarepeats R1R2 R3R4 (5 x 8) + 1 codon = 123 bp R2 CCTCATGGT GGCTCGGGGCAG prohisgly trpgln R4 CCTCATGGT GGCTCGGGTCAA prohisgly trpgln R3 CCCCATGGT GGCTCGGGACAG prohisgly trpgln R1 CCTCAGGGCGGT GGCTCGGGGCAG proglngly trpgln R2 CCTCATGGT GGCTCGGGGCAG prohisgly trpgln
 + 1 codon = 123 bp R2 CCTCATGGT GGCTCGGGGCAG prohisgly trpgln R4 CCTCATGGT GGCTCGGGTCAA prohisgly trpgln R3 CCCCATGGT GGCTCGGGACAG prohisgly trpgln R1 CCTCAGGGCGGT GGCTCGGGGCAG proglngly trpgln R2 CCTCATGGT GGCTCGGGGCAG prohisgly trpgln")
84
Normal Pathology

85
Generation of H 2 O 2 Cu 2+ + 2PrP PrP 2 -Cu Shiraishi et al Biochem J 2005;387: 247-255 H20H20 H0 ·

86
Built in of PrP Sc in lipid rafts ‘Transformation of PrP C to PrP Sc occurs preferentially in cholesterol rich lipid rafts’ ( Hooper Biochem Soc Trans 2005; 32: 335-338) ‘Cell membranes have a role in transformation of PrP C to PrP Sc ’ ( Kazlauskaite & Pinheiro Biochem Soc Symp 2005; 72: 211-222) ‘Prion replication alters the distribution of synaptophysin in membrane’ ( Russelakis- Carneiro et al Am J Pathol 2004; 165: 1839-1848)
 ‘Cell membranes have a role in transformation of PrP C to PrP Sc ’ ( Kazlauskaite & Pinheiro Biochem Soc Symp 2005; 72: ) ‘Prion replication alters the distribution of synaptophysin in membrane’ ( Russelakis- Carneiro et al Am J Pathol 2004; 165: )")
88
Protein misfolding to neuronal dysfunction pathogenesis Protein misfolding & accumulation diseases: Alzheimer’s disease Parkinson’s disease Tauopathies –Progressive Supranuclear palsy –Fronto-temporal dementias –Cortico-basal degeneration

89
Side step: Alzheimer’s disease Non infective disease Leading to dementia

90
Amyloid Precursor Protein Signal peptide Cystein rich area GlycosylationAβAβ 671-713 Phosphorylation Membrane Extra cellular daefrhdsgyevhhqklvffaedvgsnkgaiiglmvggvvia β - secretaseα - secretaseγ - secretase

91
ABeta generation

92
Toxic oligomers in ABeta Cu 2+ leads to H 2 O 2 production A Beta density in AD is related to synapse loss (lower level of synaptofysin) Small oligomers more toxic than large aggregates Sounds familiar???
 Small oligomers more toxic than large aggregates Sounds familiar")
93
Protein-normal folded Protein-abnormal folded – small aggregates (toxic) Protein aggregated (non toxic?) Protein folding diseases Mutation in protein Chaperones/Ubiquitin/ Clipping mechanisms age + - - Hydrophobic interactions + amyloid H2O2H2O2 lipid raft: synapse dysf membrane damage
 Protein aggregated (non toxic ) Protein folding diseases Mutation in protein Chaperones/Ubiquitin/ Clipping mechanisms age Hydrophobic interactions + amyloid H2O2H2O2 lipid raft: synapse dysf membrane damage")
94
Break down of referred patients April 1, 1998-December 1, 2006 1322 referred 595 ‘pending file’ 727 ‘true’ referrals 635 referrals at study end-point 332 Prion cases (279 definite + 53 probable) Mean mortality 1.09 /10 6.yr 289 non Prion (164 definite + 125 probable) 32 info refused 17 ‘pending classification’ 43 alive 13 possible prion disease
 Mean mortality 1.09 /10 6.yr 289 non Prion (164 definite probable) 32 info refused 17 ‘pending classification’ 43 alive 13 possible prion disease")
95
Incidence of CJD Deaths in Canada (per Million Population by Year) Average incidence rate of CJD in Canada = 1.09 per million Canadians 40 42 29 36 30 35 31 24 18 13 3 2 23
 Average incidence rate of CJD in Canada = 1.09 per million Canadians")
96
Classification of Referrals Definite CJD 279 Definite sporadic CJD (sCJD)256 Definite familial CJD (fCJD)5 Definite GSS11 Definite FFI1 Definite sporadic GSS1 Definite iatrogenic CJD (iCJD)4 Definite variant CJD (vCJD)1 Probable CJD 53 Probable sporadic CJD (sCJD)49 Probable familial CJD (fCJD)4 Possible CJD 13 Possible sporadic CJD (sCJD)13 Non-CJD289 TOTAL635
256 Definite familial CJD (fCJD)5 Definite GSS11 Definite FFI1 Definite sporadic GSS1 Definite iatrogenic CJD (iCJD)4 Definite variant CJD (vCJD)1 Probable CJD 53 Probable sporadic CJD (sCJD)49 Probable familial CJD (fCJD)4 Possible CJD 13 Possible sporadic CJD (sCJD)13 Non-CJD289 TOTAL635")
97
CJD Cases (n=332)
")
98
sCJD Cases by Province/Territory BRITISH COLUMBIA 40 99% ALBERTA 27 84% SASKATCH- EWAN 15 161% MANITOBA 16 144% QUEBEC 68 97% ONTARIO 115 96% NEWFOUNDLAND4 83% PRINCE EDWARD ISLAND0 NOVA SCOTIA11 124% NEW BRUNSWICK10 141% NORTHWEST TERRITORIES 0 YUKON NUNAVUT Actual sCJD % of expected CJD (based on 2006 population)
")
99
Canadian iCJD cases (dura mater) M 19 F 49 M 14 F 59 Duration of Illness Incubation Period
 M 19 F 49 M 14 F 59 Duration of Illness Incubation Period")
100
vCJD criteria in Canadian case IA Progressive neuropsychiatric disorder B Duration of illness > 6 months C No alternative diagnosis D No iatrogenic exposure IIA Early psychiatric symptoms B Painful dysaesthesias C Ataxia D Myoclonus or chorea or dystonia E Dementia IIIA No EEG/No ‘classical’* EEG *Generalized triphasic periodic complexes at approx. 1 per second B MRI pulvinar sign positive IV Positive tonsil biopsy for PrP Possible vCJD only (without pathological confirmation)
.")
102
Occipital Neocortex H&E of Canadian Case

103
Travel history Sep ’87-Aug ’90: UK education + 2 weeks visit France. Feb ’91-Mar ’91: visit UK Feb ’92-Mar ’92: visit UK June 2000: visit UK Total 38 months risk exposure 1-14 years prior to symptoms

104
duration of disease (mo) meanmedian sCJD6.3 ± 0.34 fCJD18.0 ± 6.010 GSS69.3 ± 18.635 non-CJD22.8 ± 2.316 Survival all types of CJD and non-CJD
 meanmedian sCJD6.3 ± 0.34 fCJD18.0 ± GSS69.3 ± non-CJD22.8 ± Survival all types of CJD and non-CJD")
105
Casesof which unsuspected cases sCJDDefinite25622 (8.6 %) Probable49 GSSDefinite118 ( 73 %) Probable0 fCJDDefinite50 Probable4 iCJDDefinite40 Probable0 vCJDDefinite10 Probable0 Total33230 (9.0 %)
 Probable49 GSSDefinite118 ( 73 %) Probable0 fCJDDefinite50 Probable4 iCJDDefinite40 Probable0 vCJDDefinite10 Probable0 Total33230 (9.0 %)")
106
Why where they missed? Lack of experience? Different age of onset/duration? Different clinical signs and symptoms? Different subtype? Different auxiliary investigations findings?

107
Conclusions unsuspected Prion disease cases-1 9 % of Prion disease cases were clinically unsuspected in Canada in last 8½ years. GSS poorly recognized (73% of GSS are clinically unsuspected in this series)
.")
108
sCJD and GSS can be missed clinically Missing is due to the fact that these cases are atypical: –Less usual subtypes (MM2, MV2) –Missing symptoms in the presentation –Longer duration of disease Or (occasionally) due to missed clues: MRI T2/FLAIR attenuation EEG Missing is not due to ‘academic status’ of clinician...but...are we missing much more cases?? Conclusions unsuspected Prion disease cases-2

109
‘Back of the envelope’ Minimum True Mortality rate of CJD for Canada = 1.09/10 6.yr Maximum True Mortality rate of CJD for Canada = ? General autopsy rate in Canada: 9.5 % = 1:10.5 = 1:(9.5 + 1) means 9.5x(30/332)x0.91x1.09/10 6.yr unnoticed CJD as they are not autopsied: actual CJD frequency in Canada could be max 1.09 /10 6.yr + 0.85/10 6.yr = 1.94/10 6.yr Comparison: European surveillance figures for Switzerland, Spain, Italy, France and Austria have been last 2 or more years well over 1/ 106.yr, and even over 2 in selected case. True mortality figure is probably somewhere between 1.09 and 1.94.
 means 9.5x(30/332)x0.91x1.09/10 6.yr unnoticed CJD as they are not autopsied: actual CJD frequency in Canada could be max 1.09 /10 6.yr /10 6.yr = 1.94/10 6.yr Comparison: European surveillance figures for Switzerland, Spain, Italy, France and Austria have been last 2 or more years well over 1/ 106.yr, and even over 2 in selected case. True mortality figure is probably somewhere between 1.09 and")
110
Prion diseases - regional distribution in the Netherlands until 2000 -

111
Is sporadic CJD really sporadic? Concept of spontaneous generation of abnormal prion protein is difficult to grasp But no relation with food, surgery or other possible cause found However in UK geographical clustering seen 15-25 years before onset

Similar presentations




























>")






 rare progressive neurodegenerative disorders that affect both humans and animals. They.>")



, is a fatal brain disorder that occurs in cattle.>")





>")