Download presentation
Presentation is loading. Please wait.

1
Dr A. Mousavi

2
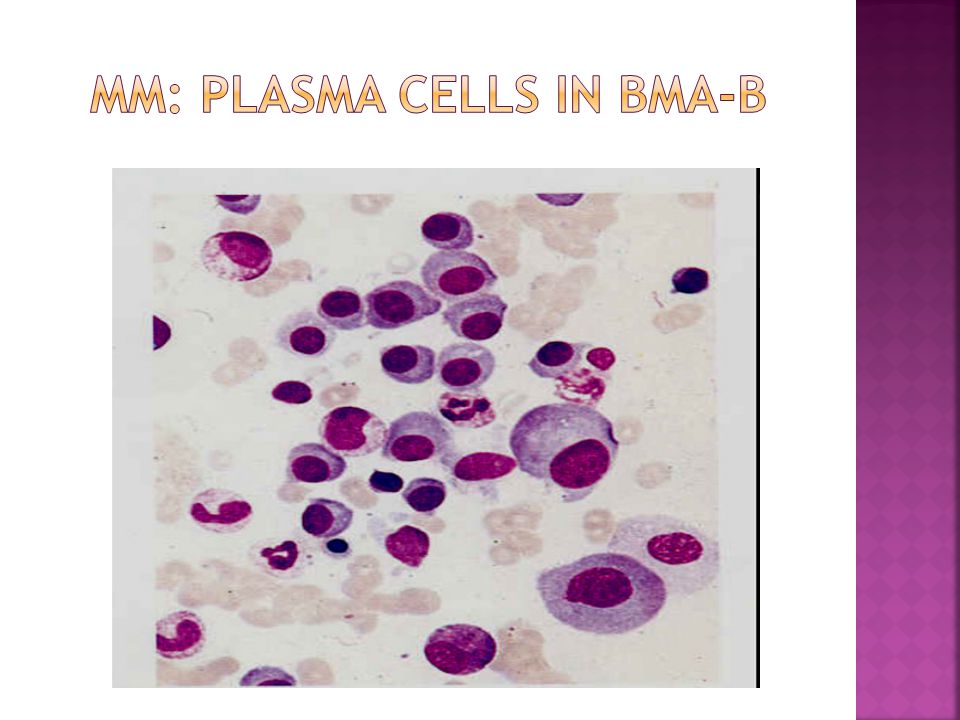
15 % of all malignant white cell diseases 1% of all cancer deaths Group of lymphoid neoplasms of terminally differentiated B-cells that have in common the expansion of a single clone of immunoglobulin(Ig)-secreting plasma cells and a resultant increase in serum levels of a single homogeneous(monoclonal) Ig or it’s fragments.
-secreting plasma cells and a resultant increase in serum levels of a single homogeneous(monoclonal) Ig or it’s fragments.")
3
Caused by malignant changes to plasma cells or the B lymphocyte cell line. Exhibit either: Excessive amounts of normal immunoglobulin proteins (Igs) Accumulation of Igs in an abnormal location Structurally abnormal Igs
 Accumulation of Igs in an abnormal location Structurally abnormal Igs.")
4
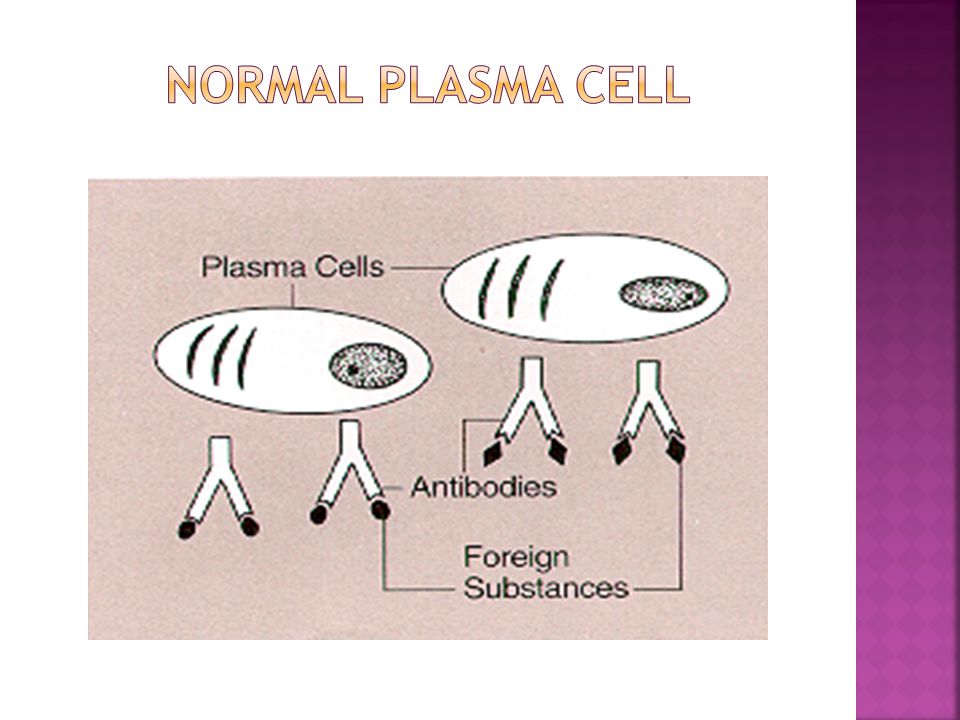
Terminally differentiated B-cells Not normally found in peripheral blood. Account for less than 3.5% of nucleated cells in the bone marrow Oval cells with low N:C ratio. Cytoplasm is basophilic blue. Nucleus (30-40% of the cell) is oval or round and typically placed eccentrically (to one side)of the cell. A clear, colorless area adjacent to the nucleus contains Golgi apparatus Russell bodies: Globules(2-3μm) of accumulated immunoglobulins in the cytoplasm of plasma cells. Usually round. Russell bodies may be found in normal bone marrow.
 is oval or round and typically placed eccentrically (to one side)of the cell. A clear, colorless area adjacent to the nucleus contains Golgi apparatus Russell bodies: Globules(2-3μm) of accumulated immunoglobulins in the cytoplasm of plasma cells. Usually round. Russell bodies may be found in normal bone marrow..")
6
Develop from stem cells in bone marrow Stem cells develop into B cells (B lymphocytes) Antigens enter body then B cells develop into plasma cells Produce antibodies
 Antigens enter body then B cells develop into plasma cells Produce antibodies")
8
Which of these are the heavy chains? Name the 5 classes of heavy chain. Gamma, mu, alpha, delta and epsilon Which of these are the light chains? Name the 2 classes of light chain. Kappa and lambda NH3+ COO-

9
Where is the constant region of the molecule? Where is the variable region? Which region defines the specificity of the antibody? Variable Which region is responsible for the physical properties of the antibody, such as ability to activate complement and binding to macrophages? Constant NH3+ COO-

11
Normally, plasma cells produce immunoglobulins to fight infection However, in MM and MGUS a single cloned plasma cell proliferate and overproduce the same Ig ("M-protein" or “Paraprotein") The M-protein is usually an IgG MM cells can also just produce the light chain component (Instead of the entire Ig)
 The M-protein is usually an IgG MM cells can also just produce the light chain component (Instead of the entire Ig)")
12
Consequence of producing lots of monoclonal Ig: o Hyperviscosity o Kidney Damage (from light chains only) o Bone pain, hypercalcemia and pathologic fractures from bone lesions. o Anemia/Pancytopenia from bone marrow invasion

13
Y B Cell Normal Plasma Cell Malignant Plasma Cell Y Y Y Y Y Y Y Y Y Y Y Y Y Y Y Y Y Y Y Y Y Y Y Y Y

14
Accumulation of a single protein that arises from proliferation of a single plasma cell clone. Since each B cell can respond to only one antigenic epitope, a plasma cell derived from that B cell produces antibody that is reactive against that unique epitope (monoclonal antibody). Malignant changes to that plasma cell result in uncontrolled production of its specific antibody. The specificity of the monoclonal antibody (M protein) varies between patients, but each affected patient has only one M protein specificity.
. Malignant changes to that plasma cell result in uncontrolled production of its specific antibody. The specificity of the monoclonal antibody (M protein) varies between patients, but each affected patient has only one M protein specificity..")
15
Adhesion molecules Stromal cells Interactions: Cytokines (IL-6) Growth factors that promote angiogenesis (IGF-1, VEGF, SDF-1α) Inactivated immune system
 Growth factors that promote angiogenesis (IGF-1, VEGF, SDF-1α) Inactivated immune system")
16
Gammopathy Monoclonal gammopathy Dysproteinemia Paraproteinemia

17
MGUS (62%) Malignant Monoclonal gammopathies: MM (18%) variants: smoldering myeloma (3%), non-secretory MM, light chain myeloma Plasmacytoma Plasma cell leukemia IgD myeloma POEMS syndromes (Osteosclerotic myeloma) Waldenstrom’s Macroglobulinemia (Lymphoplasmacytic lymphoma)
 Malignant Monoclonal gammopathies: MM (18%) variants: smoldering myeloma (3%), non-secretory MM, light chain myeloma Plasmacytoma Plasma cell leukemia IgD myeloma POEMS syndromes (Osteosclerotic myeloma) Waldenstrom’s Macroglobulinemia (Lymphoplasmacytic lymphoma)")
18
Malignant lymphoproliferative disorders Heavy chain diseases (Gamma HCD, Mu HCD, Alpha HCD) Immunoglobulin deposition diseases (Primary Amyloidosis, Systemic light chain and heavy chain deposition diseases)
 Immunoglobulin deposition diseases (Primary Amyloidosis, Systemic light chain and heavy chain deposition diseases)")
19
In any suspected Monoclonal Gammopathy should include to accurately classify the disorder: Complete Blood Count (look for anemia) Comprehensive Metabolic panel Look for renal insufficiency, hypercalcemia and subtle clues like decreased anion gap
 Comprehensive Metabolic panel Look for renal insufficiency, hypercalcemia and subtle clues like decreased anion gap")
20
Total protein and albumin level. Determine Globulin component.Too low globulin( 3.5gm%) is concerning:Determine if Polyclonal vs.Monoclonal. Evaluate further with: Quantitative Immunoglobulins: Increase in all components usually, polyclonal. Increase in single component with reciprocal decrease of uninvolved globulin usually, may suggest monoclonal.
 is concerning:Determine if Polyclonal vs.Monoclonal. Evaluate further with: Quantitative Immunoglobulins: Increase in all components usually, polyclonal. Increase in single component with reciprocal decrease of uninvolved globulin usually, may suggest monoclonal..")
21
Serum Protein Electrophoresis with immunofixation if monoclonal gammopathy is suspected. 24HR-Urine protein electrophoresis with urine immunofixation (Serum Free Light Chain assay(κ/λratio) may be used in place of UPEP}
 may be used in place of UPEP}.")
22
Bone marrow biopsy to evaluate% plasma cells if there is monoclonal protein or abnormal UPEP or Light chain assay or if strong clinical picture of myeloma. Skeletal survey if monoclonal gammopathy has been established(Bone scans are usually negative in MM) Beta-2 microglobulin and Albumin for staging and prognosis in MM (once diagnosis is made).
 Beta-2 microglobulin and Albumin for staging and prognosis in MM (once diagnosis is made)..")
25
Denotes presence of an M-protein in a patient without a plasma cell or lymphoproliferative disorder (i.e; Undetermined Significance) M- protein < 3 gr/dl < 10% plasma cell in bone marrow No or small amounts of M- protein in urine Absence of lytic bone lesions, anemia, hypercalcemia, renal insufficiency No evidence of B- cell lymphoproliferative disorder Stability of M- protein over time
 M- protein < 3 gr/dl < 10% plasma cell in bone marrow No or small amounts of M- protein in urine Absence of lytic bone lesions, anemia, hypercalcemia, renal insufficiency No evidence of B- cell lymphoproliferative disorder Stability of M- protein over time")
26
Incidence increases with age Significance: can progress to monoclonal disease IgA or IgG MGUS IgM MGUS MM Primary Amyloidosis Related Plasma cell disorder NHL CLL Waldenstroms’ Macroglobulinemia

27
Smoldering Myeloma: Serum monoclonal protein ≥ 3 g/dl and/or bone marrow plasma cells ≥ 10% No end organ damage related to plasma cell dyscrasia

30
Rare variant: About 1% of Myelomas May present with Bone lesions (most common presenting symptom bone pain) No serum or urine monoclonal protein(diagnosis can be missed if one is not aware of this entity, NSMM). Renal failure and hypercalcemia are generally lacking Anemia may be present Bone marrow biopsy must be performed in suspected cases: Immunostaining for a monoclonal protein on bone marrow sections may establish the diagnosis Clonal plasma cell population in marrow. Must rule out IgD and IgE myeloma

31
Localized plasma cell tumor Absence of a plasma cell infiltrate in random marrow biopsies No evidence of other bone lesions by radiographic examination Absence of renal failure, hypercalcemia or anemia Younger median age at presentation (55 y) Treatment: Radiation to site (5000 cGy) 50-60% will convert MM within 10 years Possible bone marrow collection/ storage
 Treatment: Radiation to site (5000 cGy) 50-60% will convert MM within 10 years Possible bone marrow collection/ storage")
32
Arise outside the bone marrow with no features of MM Most common: Head and Neck region Less common: Lymph nodes, Salivary glands, spleen, liver,… 25% have small monoclonal spike Rare dissemination, rare revolution to myeloma Management: If completely resected during biopsy,no further therapy If incompletely resected,radiation therapy locally

33
Three criteria: 1.Presence of a serum or urinary monoclonal protein 2.Presence of 10 percent or more clonal plasma cells in the bone marrow or a plasmacytoma 3.Presence of end organ damage felt related to the plasma cell dyscrasia, such as: M- CRAB: Monoclonal protein HyperCalcemia (calcium >11.5 gm/dl) Renal Insufficiency Anemia (Hb < 10gm/dl) Lytic Bone lesions
 Renal Insufficiency Anemia (Hb < 10gm/dl) Lytic Bone lesions")
34
Bone lesions: Conventional Radiographs (Skeletal Survey) is abnormal in 80% of MM Focal lytic bone: 57% Osteopenia or Osteoporosis: 20% Pathologic fractures: 20% Vertebral body compression fractures: 20%
 is abnormal in 80% of MM Focal lytic bone: 57% Osteopenia or Osteoporosis: 20% Pathologic fractures: 20% Vertebral body compression fractures: 20%")
36
Anemia: Normochrome nomocyter in 75% of MM Hb < 10 mg/dl Renal insufficiency: Serum creatinine increased in >50% at diagnosis Creatinine >2g/dL in 20% of patients Renal failure may be presenting manifestation

37
Cytogenetic: 14 q 32 1 q 5, 8, 12,…. Deletion 17 p and Abnormalities associated with chromosome13 carry a particularly unfavorable prognosis and respond poorly to therapy

38
Staging: International staging system: Stage I— B2M <3.5mg/L and serum albumin ≥ 3.5g/dL Stage II— neither stage I nor stage III Stage III— B2M ≥ 5.5mg/L Median overall survival dicreases with increasing stages

39
Indications: presence of any of CRAB, High risk patients Current frontline options: Conventional chemotherapy Survival ≤ 3 yrs Stem Cell Transplantation Prolongs survival 4- 5 yrs Novel agents targeting stromal interactions and associated signaling pathways (Thalidomide, Lenalidomide, Bortezomib,…) have shown promise and improved survival
 have shown promise and improved survival")
40
P olyneuropathy (Motor, 100%) O rganomegaly (Hepatosplenomegaly, 50%) E ndocrinopathy (Hypogonadism, Hypothyroidism, 66%) M onoclonal gammopathy S kin changes (Hyperpigmentation, Hypertrichosis) S clerotic bone lesions (related to cytokines, VEGF, 97%)
 O rganomegaly (Hepatosplenomegaly, 50%) E ndocrinopathy (Hypogonadism, Hypothyroidism, 66%) M onoclonal gammopathy S kin changes (Hyperpigmentation, Hypertrichosis) S clerotic bone lesions (related to cytokines, VEGF, 97%)")
41
Diagnostic criteria: Two major+ at least one minor Major: Polyneuropathy, Monoclonal plasma cell disorder Minor: Sclerotic bone lesions, Organomegaly, castleman’s dis, Volume overload, Endocrinopathy, Skin changes, Papilledema Treatment: Radiation to bone lesion

42
>2 X 10 9 /L plasma cells in blood(seen on peripheral smear) Younger age Higher incidence of organomegaly and lymphadenopathy More extensive bone marrow infiltration Renal failure more common Less bone pain, fewer lytic lesions Poor response to therapy
 Younger age Higher incidence of organomegaly and lymphadenopathy More extensive bone marrow infiltration Renal failure more common Less bone pain, fewer lytic lesions Poor response to therapy")
44
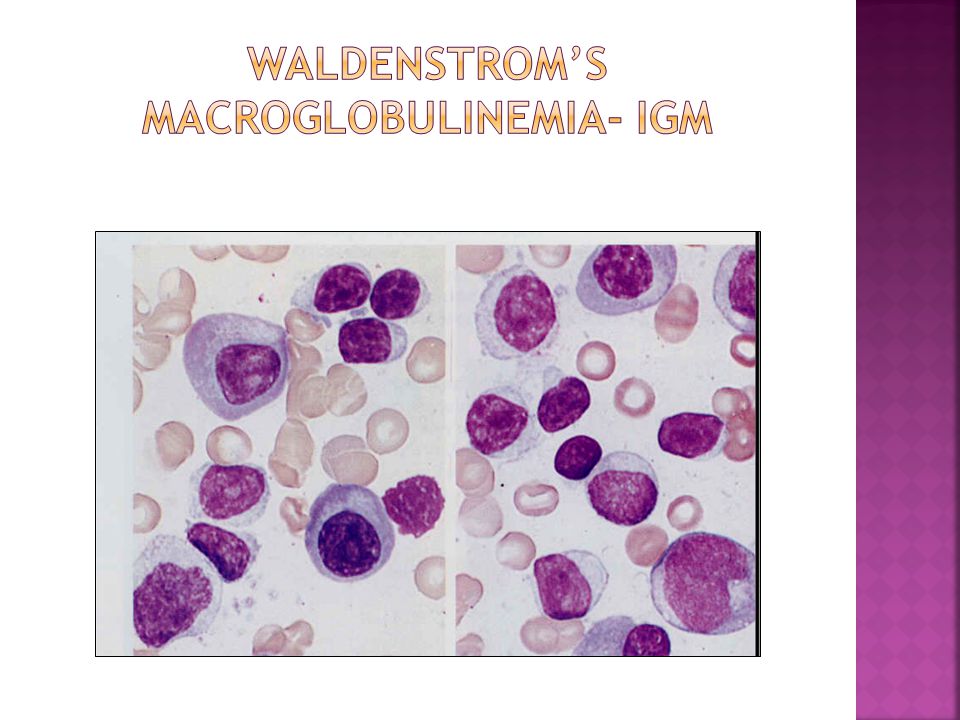
Monoclonal gammopathy: IgM type Plasmacytoid lymphoma Median age at diagnosis: 60 yrs Presentation: Hyperviscosity syndrome(15%): visual impairment, neurologic manifestations Bleeding(Acquired VWD) Cryoglobulinaemia Organomegaly, lymphadenopathy (20%- 40%) Autoimmune hemolysis: common Bone marrow involvement: 90% Lytic bone lesions: 2% Hypercalcemia: 4%
: visual impairment, neurologic manifestations Bleeding(Acquired VWD) Cryoglobulinaemia Organomegaly, lymphadenopathy (20%- 40%) Autoimmune hemolysis: common Bone marrow involvement: 90% Lytic bone lesions: 2% Hypercalcemia: 4%")
46
Asymptomatic patients not treated until symptoms develop If Hyperviscocity features: urgent Plasmapheresis Symptomatic WM: Rituximab based therapy

47
Amyloidosis caused by extracellular deposition of pathologic insoluble fibrillar proteins in organs and tissues M-protein: 89%, Lambda: 70% M-protein > 3 mg/dl: 7% Hypogammaglobulinemia: 20% Median bone marrow plasma cells: 7% (<10%) Organ involvments: cardiac arrythmia, renal failure, skin changes, macroglossia
 Organ involvments: cardiac arrythmia, renal failure, skin changes, macroglossia")
48
Evaluate for amyloidosis in patients with a monoclonal protein in serum or with a monoclonal protein in serum or urine plus: Nephrotic syndrome or renal insufficiency Congestive heart failure Peripheral neuropathy Carpal tunnel syndrome Hepatomegaly Idiopathic malabsorption

49
Diagnostic Criteria: Tissue biopsy showing typical morphology Apple green birefringence under polarized light after Congo Red staining Term amyloid first coined by Virchow in mid 19 th century (meaning starch or cellulose). Typical fibrillar ultrastructure Diagnostic methods and Sensitivity: Bone marrow examination: 56% Abdominal fat aspiration: 80% Combined BM and fat aspirate: 89%

52
Heavy chain of Ig Alpha type: Younger patients Mediterranean lymphoma (Intestinal lymphoplasmacytoid Lymphoma) Remission with antibiotics
 Remission with antibiotics")
53
Gamma Heavy Chain Disease – seen in elderly Symptoms –enlarged liver and spleen, recurrent infections, and anemia Some patients experience no symptoms Treatment with anti-lymphoma drugs and corticosteroids Mu Heavy Chain Disease – rare Symptoms include enlarged spleen, liver and abdominal lymph nodes Survival and response to treatment varies

55
Thank you for your attention

Similar presentations










 Family Medicine Review Course 2011 Christian Cable, MD, FACP.>")





 Idiopathic Associated with other diseases (autoimmune, infectious, non-heme.>")








>")

BB Topic 3 Autoimmunity Part 8 Immunoproliferative Diseases.>")

