Download presentation
Presentation is loading. Please wait.

1
Investigation Of Hemoglobinopathy. Dr Mary Ann Anderson Hematology Laboratory Registrar Scientific Meeting 0730 am 19/6/08.

2
Introduction. ► Haemoglobinopathies are ‘inherited abnormalities of globin chain synthesis’ (B. Bain 2007). ► Encompass a clinical spectrum from asymptomatic findings on blood film to death in utero. ► Wide spread distribution throughout the world and increasing prevalence in multicultural Australia.
. ► Encompass a clinical spectrum from asymptomatic findings on blood film to death in utero. ► Wide spread distribution throughout the world and increasing prevalence in multicultural Australia..")
3
Hemoglobin Structure. ► Hemoglobin transports oxygen to the tissues. ► Each RBC contains hemoglobin. ► A normal hemoglobin molecule consists of: Four globin chains (2 alpha, 2 beta). Each globin chain has an iron containing heme molecule. The iron in the heme molecule binds to oxygen.
. Each globin chain has an iron containing heme molecule. The iron in the heme molecule binds to oxygen..")
5
Genetics. ► In the first 8 weeks of embryonic life the predominant forms of hemoglobin are: Hb Gower 1 (ζ 2 ε 2 ). Hb Gower 2 (α 2 ε 2 ). Hb Portland 1 (ζ 2 γ 2 ). ► By the 12 th week embryonic hemoglobin is replaced by Hb F (α 2 γ 2 ) which represents 70 – 100% of hemoglobin in fetal life.
. Hb Gower 2 (α 2 ε 2 ). Hb Portland 1 (ζ 2 γ 2 ). ► By the 12 th week embryonic hemoglobin is replaced by Hb F (α 2 γ 2 ) which represents 70 – 100% of hemoglobin in fetal life..")
6
Genetics (2). ► Adult hemoglobin Hb A (α 2 β 2 ) detectable from 16/40, replaces Hb F as predominant hemoglobin by 6/12 after birth, up to 30% of Hb in fetal life. ► Hemoglobin HbA2 (α 2 Δ 2 ) is present in utero but only very minor in normal adults. ► In normal adults 96 – 98% of hemoglobin is HbA, Hb A2 (2 – 3%) and HbF (<1%) constitute a minor component of the total hemoglobin.
 detectable from 16/40, replaces Hb F as predominant hemoglobin by 6/12 after birth, up to 30% of Hb in fetal life. ► Hemoglobin HbA2 (α 2 Δ 2 ) is present in utero but only very minor in normal adults. ► In normal adults 96 – 98% of hemoglobin is HbA, Hb A2 (2 – 3%) and HbF (<1%) constitute a minor component of the total hemoglobin..")
7
Copyright ©1997 BMJ Publishing Group Ltd.

9
α2Δ2α2Δ2 α2γ2α2γ2

10
Geography. ► Commonest genetic defect world wide with an estimated 269 million carriers. ► 90 million carriers in South East Asia, 85 million in Sub Saraharan Africa, and 48 million in the West Pacific region. ► Distributed across South East Asia in a line stretching from Southern China down the Malaysian Peninsula to Indonesian islands.

11
Geography (2). ► Also distributed across the Mediterranean, Middle East, and Indian Subcontinent. ► The distribution of the defect is thought to be due to partial protection for carriers from Plasmodium Falciparum Malaria.

14
Beta thal prevalence.

15
Haemoglobinopathy Classification. ► Thalassaemia: Primary abnormality is a reduced rate of synthesis of globin chains. Defined by imbalance αβ ratio. Traditionally though not invariably microcytic hypochromic anemia. ► Variant hemoglobin's in which there is a structural abnormality in the globin chain.

17
Alpha Thalassaemia Classification. ► Generally caused by gene deletions. ► α + chromosome produces some α globin (eg - α/- α). ► α 0 chromosome produces no α globin (eg - -/ α α).
. ► α 0 chromosome produces no α globin (eg - -/ α α)..")
18
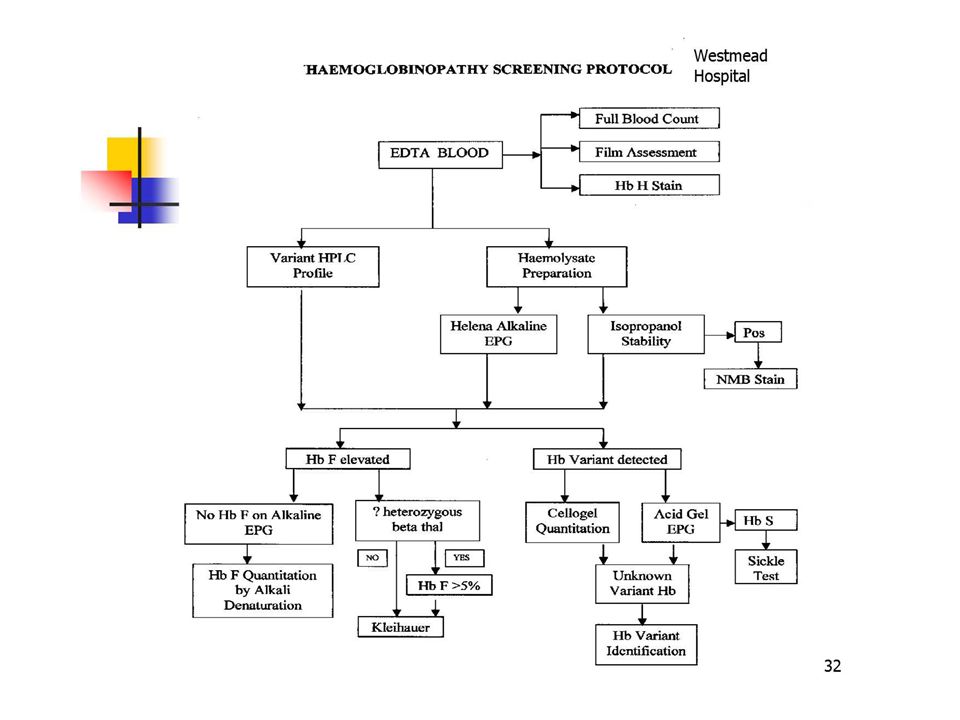
Alpha Thalassaemia Classification (2). ► α trait due to deletion of one or two of the four alpha genes, asymptomatic (eg - α/ α α, --/ α α, - α/- α). ► Hemoglobin H disease is the lack of three of the four α genes resulting in alpha thalassaemia major. ► Hemoglobin Bart’s Hydrops Fetalis results from absence of all four α genes, incompatible with post natal life.
. ► Hemoglobin H disease is the lack of three of the four α genes resulting in alpha thalassaemia major. ► Hemoglobin Bart’s Hydrops Fetalis results from absence of all four α genes, incompatible with post natal life..")
19
Beta Thalassemia Classification. ► β thal major is homozygosity or compound heterozygosity resulting in severe phenotype. ► β thal minor or trait is heterozygosity with asymptomatic phenotype. ► β thal intermedia is an intermediate phenotype produced by a variety of genotypes.

20
Beta Thalassemia Classification (2). ► β 0 syndromes are characterized by the affected gene producing no beta chain. ► β + syndromes are characterized by the abnormal gene producing beta chains at a reduced rate. ► Usually due to point mutations.

21
Variant Hemoglobin Classification. ► The variant haemoglobins are disorders of globin chain synthesis. ► Normal αβ ratio so most have normal MCV and MCH. ► There are over 1000 mutations associated with the haemoglobinopathies most of which will produce variants.

22
Variant Hemoglobin Classification. ► Initially recognized forms were classified alphabetically (Hb C, D, E), subsequent naming after the location of discovery. ► The most common forms in Australia include Hb S, Hb E, Hb Constant Spring and Hb C. ► Usually caused by point mutations.
, subsequent naming after the location of discovery. ► The most common forms in Australia include Hb S, Hb E, Hb Constant Spring and Hb C. ► Usually caused by point mutations..")
23
FBC. ► Thalassaemias are typically microcytic and hypochromic anemia's. ► Thalassemia causes a uniform microcytosis without increase in RDW (cf iron deficiency). ► Hb H and Δβ thal however can cause an increased RDW.
. ► Hb H and Δβ thal however can cause an increased RDW..")
24
FBC (2). ► RBC often increased in thal but decreased in iron deficiency and AOCD. ► Hb typically normal in thal minor but decreased in intermedia and major syndromes. ► MCV is the most valuable parameter in predicting thal.

25
Alpha Thalassaemia Major - target cells, microcytosis, hypochromia, NRBCs, poikilocytes, polychromasia, basophilic stipling, tear drops.

26
βThal Major anisocytosis, poikilocytosis, targets, tear drops, fragments, hypochromaia, basophilic stippling.

27
Iron Studies. ► Except in the urgent situation of pregnancy iron deficiency should always be excluded and treated prior to work up for thal. ► MCV and MCH are influenced by iron deficiency. ► Hb A2 can be lowered by iron deficiency falsely normal results if tested when iron deficient (false negatives).
..")
28
Table 1: Laboratory features in different clinical states. Iron Deficiency Chronic Disease Iron Overload Thalassemia Haemoglobin N or ↓↓N Serum Fe↓↓↑ ↑ or N Transferrin Receptor ↑ ↓ or N ↓N Transferrin Sat.↓↓↑N Ferritin↓↑or N↑ MCV↓ ↓ or N N↓ Marrow Fe↓↑↑↑

29
Hb H Inclusions. ► Hb H is an insoluble tetramer consisting of four beta globin chains, due to a lack of alpha chains in alpha thal major. ► Oxidation of these tetramers provokes precipitation which can be visualized microscopically as ‘golf ball’ inclusions. ► Oxidation can be precipitated by oxidative dyes (although there is significant batch to batch variability making controls essential).
..")
30
Hb H Inclusions (2). ► In Hb H disease 30 – 100% of RBCS contain Hb H inclusions. ► In alpha thal minor there is one cell with Hb H inclusions per 1000 – 10,000 RBCS. ► Other nucleic acid and protein precipitates also stain (without ‘golf ball’ pattern).
..")
31
Hb H inclusions (3). ► When there is a reticulocytosis a rare Hb H inclusion may be missed – operator experience crucial. ► Detection of Hb H inclusions points to an alpha chain mutation and narrows the amount of DNA analysis required. ► False negatives problematic, even with 2 alpha gene deletions no inclusions may be seen after several minutes of searching.

33
Kliehauer Test For Hb F. ► Used to detect the presence of Hb F (fetal hemoglobin). ► RBCS on a slide are stained to detect the presence of Hb F. ► Can distinguish hetrocellular HbF from pancellular HbF seen in HPFH.

34
Kleihauer Test For Hb F (2). ► Rarely done and difficult to interpret and standardize due to significant variability between observers. ► Confirms maternal blood contamination with fetal blood in cases of fetomaternal hemorrhage, with D mismatch. ► Flow cytometry is now the primary tool for investigation of fetal haemoglobins in Australia.

36
Electrophoresis Principle. ► Separation of haemoglobins with electrophoresis at pH 8.4 (alkaline) and pH 6.2 (acid). ► Scanning allows quantification of the hemoglobin present, bands are seen by staining. ► At alkaline pH Hb C, E, A2 and O migrate together to form a single band, Hb S, D and G also co migrate.
 and pH 6.2 (acid). ► Scanning allows quantification of the hemoglobin present, bands are seen by staining. ► At alkaline pH Hb C, E, A2 and O migrate together to form a single band, Hb S, D and G also co migrate..")
37
Electrophoresis Principle (2). ► At acid pH Hb C separates from E and O and Hb S separates from D and G. ► Hb E and O cannot be separated by electrophoresis neither can Hb D and G.

38
(1) Normal (2) New born (3) Hb C trait [A-C] (4) Hb SC disease [S-C] (5) Sickle cell disease [S-S], (6) Sickle cell trait [A – S] (7) New born (8) Normal.
![(1) Normal (2) New born (3) Hb C trait [A-C] (4) Hb SC disease [S-C] (5) Sickle cell disease [S-S], (6) Sickle cell trait [A – S] (7) New born (8) Normal.](http://images.slideplayer.com/12/3382077/slides/slide_38.jpg "(1) Normal (2) New born (3) Hb C trait [A-C] (4) Hb SC disease [S-C] (5) Sickle cell disease [S-S], (6) Sickle cell trait [A – S] (7) New born (8) Normal.")
39
Electrophoresis Interpretation. HbA2 range Interpretation > 7.0 % Rare, repeat to verify test. Exclude a structural variant. Can be due to rare β thal mutations. 3.8 – 7.0 % Beta thal trait or unstable Haemoglobin. 3.4 – 3.7 % Fe deficiency in β thal trait; Δ chain variant with β thal trait. Interaction of α and β thal traits; rare β thal mutations. HbS making measurement inaccurate; interaction of α - Hb S. 2.0 – 3.3 % Normal. Δ and β thal (but HbF should be elevated); alpha thal trait. Rare cases of β thal trait coexisting with either Δ or α thal trait. < 2.0 % Δ β thal (but HbF should be elevated). Alpha thal trait; Hb H disease; Δ variant or delta Thalassemia. Iron deficiency.
; alpha thal trait. Rare cases of β thal trait coexisting with either Δ or α thal trait. < 2.0 % Δ β thal (but HbF should be elevated). Alpha thal trait; Hb H disease; Δ variant or delta Thalassemia. Iron deficiency..")
40
Electrophoresis Strengths. ► Commercial, widely available, rapid methods used for many years. ► Gives an estimate of HbA2 level. ► Identifies some variant haemoglobins which are well characterized.

41
Electrophoresis Disadvantages. ► ► Labor-intensive. ► ► Inaccurate in quantification of low- concentration variants (HbA2) and in detection of fast variants (HbH, Hb Barts). ► ► The precision and accuracy for Hb A2 using scanning of electrophoretic gels is poor (in comparison to HPLC).
 and in detection of fast variants (HbH, Hb Barts). ► ► The precision and accuracy for Hb A2 using scanning of electrophoretic gels is poor (in comparison to HPLC)..")
42
Electrophoresis Disadvantages (2). ► ► Coefficient of variation (CV) 33.6% for gel electrophoresis (mean HbA2 concentration 2.41%). ► ► Column chromatography has CV 14.6% (mean HbA2 3.21%) and HPLC has CV 4.3% (mean HbA2 3.47%). ► ► An imprecise test in comparison to other tests now available.
 33.6% for gel electrophoresis (mean HbA2 concentration 2.41%). ► ► Column chromatography has CV 14.6% (mean HbA2 3.21%) and HPLC has CV 4.3% (mean HbA2 3.47%). ► ► An imprecise test in comparison to other tests now available..")
43
Isoelectric Focusing. ► ► Equilibrium process in which Hb migrates in a pH gradient to a position of 0 net charge can be used to separate and quantify Hb. ► ► Excellent resolution allowing precise and accurate Hb quantification. ► ► Labor-intensive and time-consuming.

44
Isoelectric Focusing (2). ► ► The migration order is the same as with alkaline electrophoreses however HbC and E separate as do HbO and S and HbD and G. ► ► Hb A and F are also clearly separated. ► ► Both more accurate and more precise than standard electrophoresis.

45
Capillary Isoelectric Focusing. ► ► Hybrid technique combining capillary electrophoresis sensitivity with automated sampling and data acquisition of HPLC. ► ► Established role in the detection and quantification of Hb variants. ► ► Separation of Hb in this method is related to the isoelectric point of the Hb, and this may enhance inter laboratory reproducibility.

46
Capillary Isoelectric Focusing (2). ► ► Used to quantify Hb variants, HbA2 and HbF. ► ► Quantitative and qualitative Hb variant results from CIEF and cation exchange chromatography are highly correlated. ► ► CIEF gives slightly better resolution of the unusual variants HbC Harlem and HbD Punjab compared to chromatography.

47
HPLC Principle. ► ► Cation-exchange HPLC can be preformed on an automated instrument that can quantify Hb A2, Hb F, Hb A, Hb S, and Hb C. ► ► Studies show equivalence or superiority over electrophoresis in terms of identification of variant haemoglobins and quantification of HbA2 level. ► ► Negatively charged carboxyl molecules bound to silica make up the cartridge matrix.

48
HPLC Principle (2). ► ► Positively charge molecules (salt and hemoglobin) bind to the carboxyl groups. ► ► Haemoglobin molecules are bound and displaced by increasing salt concentration. ► ► Haemoglobin variants separate out due to variation in charge.
 bind to the carboxyl groups. ► ► Haemoglobin molecules are bound and displaced by increasing salt concentration. ► ► Haemoglobin variants separate out due to variation in charge..")
54
HPLC Disadvantages. ► ► Hbs may co-elute or may elute before instrument peak integration. ► ► HbE, HbOsu Christianbourg, and HbG Copenhagen co-elute with Hb A2, making quantification impossible when these variants present. ► ► The measurement of Hb A2 is complicated in individuals with Hb S because the Hb A2 is falsely increased by the presence of Hb S adducts.

55
HPLC Disadvantages (2). ► ► Capillary zone Electrophoretic method can be used to quantify Hb A2 in the presence of Hb S by eliminating interference from these adducts. ► ► Interference can also be eliminated by the use of micro anion-exchange column methodology. ► ► Integration errors can result in false decreases in the values obtained, although this can be minimized by applying known corrections.

56
HPLC Disadvantages (3). ► ► Haemoglobinopathies cause inappropriately high HbA1c (HbNiigata, HbSherwood Forest, HbRambam, HbRaleigh). ► ► The presence of a structural Hb variant may adversely affect the measurement of HbA1C.
. ► ► The presence of a structural Hb variant may adversely affect the measurement of HbA1C..")
57
HPLC Strengths. ► ► Method of choice for screening for Hb variants; for quantification of HbA2 + HbF concentrations and mandatory in neonatal screening. ► ► Quicker and more sensitive than standard techniques for detecting HbF (in diagnosis of HPFH and monitoring sickle cell anemia). ► ► Indeed alkaline gel electrophoresis cannot detect HbF in healthy adults or those with marginally increased Hb F.
. ► ► Indeed alkaline gel electrophoresis cannot detect HbF in healthy adults or those with marginally increased Hb F..")
58
HPLC Strengths (2). ► ► Can be used to characterize rare haemoglobinopathies not well detected with other methods (HbRambam). ► ► Established role in the diagnosis of thalassaemia and haemoglobinopathies, including with cord blood samples.
. ► ► Established role in the diagnosis of thalassaemia and haemoglobinopathies, including with cord blood samples..")
59
HPLC Strengths (3). ► ► HPLC should be the primary method for detecting variant hemoglobin's and simultaneous quantitation of HbA2 and HbF. ► ► This replaces three separate methods: Haemoglobin electrophoresis. Quantitation of HbA2. Quantification of HbF).
..")
61
DNA Analysis. ► ► Indicated when the hemoglobinopathy not confirmed by other methods or when the underlying mutation important to management. ► ► Other techniques lead to a presumptive identification of the hemoglobinopathy only. ► ► For genetic counseling defining the particular mutation or deletion is often required – this is achieved by a variety of molecular techniques.

62
DNA Analysis (2). ► ► DNA from WBCs, amniocytes, or chorionic tissue may be utilized for diagnosis of various α and β globin chain abnormalities. ► ► Southern blot hybridization using restriction enzymes digesting labeled complementary probes define deletional mutations in α and rare β thal. ► ► PCR amplifies globin genes and utilizes allele specific primers to detect known globin chain mutations eg HbS, E, D, O + several β thal.

63
DNA Analysis (3). ► ► PCR can be used to detect unknown mutations. ► ► Aims to separate amplified DNA on gels or with HPLC on the principle that different amino acids migrate differently. ► ► 3 primary methods – mutation analysis, DNA scanning and DNA sequencing.

64
Mutation Analysis. ► ► DNA testing for thal is tailored to prevalent local mutations and suggested mutations on the basis of preliminary testing. ► ► Based on PCR which provides rapid, accurate identification of multiple single point mutations.

65
Mutation Analysis (2). ► ► In HK there are 15 common non deletional alpha gene defects and 23 common beta thal mutations. ► ► HK mutations involve single base mutations and can be simultaneously tested for by printing relevant oligonucleoties onto slides. ► ► This allows for mass screening.

66
DNA Scanning. ► ► Useful when screening large DNA segments or exons for nucleotide changes is necessary due to the possibility of many different mutations. ► ► Can dissect a gene into discrete fragments eg 500 bp in size allowing mutations to be found in large genes. ► ► Uses techniques such as SSCP (single stranded conformation polymorphism) and DGGE (denaturing gradient electrophoresis).
 and DGGE (denaturing gradient electrophoresis)..")
67
DNA Scanning (2). ► ► Denaturing HPLC is most common and based on different melting temps of hetroduplexes and homoduplexes which can be separated by EP. ► ► Any putative mutations must be confirmed by DNA sequencing to distinguish mutations from neutral polymorphisms. ► ► Hemoglobin genes are relatively small (1.6 kb with 3 exons) and DNA sequencing is increasingly accessible hence scanning is less frequently used.
 and DNA sequencing is increasingly accessible hence scanning is less frequently used..")
68
DNA Sequencing. ► ► DNA sequencing is now standard practice for looking for mutations in the beta and alpha globin genes. ► ► Indicated if mutations are not detectable with the preliminary screening and in difficult cases eg N HbA2 beta thal or silent beta thalassaemia. ► ► Difficult cases best delineated by direct gene sequencing because a number of causative mutations result in the observed phenotype.

69
DNA Sequencing (2). ► ► In beta thalassaemia there can be normal HbA2 so if the mutation absolutely needs to be excluded, DNA sequencing is preferred. ► ► Entire sequence of both α genes and most of the β globin gene can be sequenced with four primer sets. ► ► Dye primers or fluorescently labeled M13 primers are used to initiate elongation.

70
DNA Sequencing (3). ► ► In heterozygous thal DNA sequence will show a mutated gene and normal nucleotide base easy to miss the mutated gene. ► ► Overcome by sequencing forward and reverse strands but still necessary to visually inspect the sequence tedious and source of error. ► ► Becoming cheaper and more accessible, as software is developed to assist in sequence analysis.

71
DNA sequencing trace (a) forward primer, (b) reverse primer. In the reverse primer it is clear on visual inspection that there is a point mutation with both G and C being present. The forward sequence, when it has been magnified, shows a small “blip” representing the mutant C base under the normal G sequence.

72
Sickle Solubility Tests. ► Works on principle that HbS is insoluble in deoxygenated state forming crystals that refract light and cause the solution to be turbid. ► Detects HbS at conc. > 20% and differentiates HbD and G which migrate with Hb S on cellulose acetate electrophoresis at alkaline pH. ► Positive results are also obtained on samples containing both HbS and beta globulin mutations.

73
Sickle Solubility Tests (2). ► False positives can occur in leukocytosis, hyperprotienemia and unstable hemoglobin states. ► False negatives can occur in patients with anemia or if outdated buffer is used and in infants less than 6 months. ► All results must be confirmed by the more accurate HbEP or HPLC.

75
Immunoassay For Variant Hemoglobin. ► Kits are available for the immunoassay of HbS, C, E and A. ► Detect the appropriate hemoglobin down to 5 – 10%. ► These cards however can be unreliable with intermittent failure of the method.

78
Screening And Prevention Programs. ► UK guidelines: “selective testing of parents for haemoglobinopathies can be done if the percentage of patients at risk is low” ► However this policy is reliant on reliable information regarding ethnic origin being available. ► It is not always possible to predict prenatally which fetus will have beta thal major and which will have beta thal intermedia.

79
Arguments In Favor Of Universal Antenatal Screening. ► Gets around the problem of inaccurate ethnic histories. ► Minimizes the chance of a child being born with a major phenotype. ► Picks up those missed due to normal indices and normal HbA2.

80
Arguments Against Universal Antenatal Screening. ► Cost efficacy. ► Once a woman is pregnant the only way of preventing the outcome is abortion. ► Low true positive rate in our community, false positives may cause unnecessary anxiety.

81
What We Do At Alfred. ► Patients selected for HPLC based on: Physician request eg family history, microcytic, hypochromic indices. Antenatal screening clinic: ► Lab registrars review ethnicity, MCV, MCH, MCHC, film, Hb and Fer for all antenatal patients referred. ► High risk patients and their partners are referred for HPLC. ► Depending on the findings of HPLC samples may undergo further DNA analysis to characterize the mutation. ► Currently approximately 30 – 40 samples tested per month in this way.

82
Future Directions. ► The FBE/ film and ethnicity are central to screening however as the community ethnic profile changes more people will be screened. ► The UK recently moved to universal perinatal screening in this scenario HPLC is the only way to provide accurate and efficient screening. ► If an abnormal hemoglobin is deemed probable further characterization of the genetic defect with DNA techniques can be implemented.

83
Conclusions. ► Hemoglobinopathy is an important cause of disease world wide with significant implications for genetic counseling. ► An increasing number of individuals are at risk and ethnic history is unreliable, prompting moves to universal antenatal haemoglobinopathy screening. ► Increasing number of tests being preformed necessitates the use of fast, accurate and efficient HPLC with DNA analysis for further clarification.

84
Any Questions? Thank you.

Similar presentations



















 Korean J Hematol 46: 41-44 Microcytic.>")





 Hb is found in RBCs its main function is to transport O2 to tissues. Structure: 2 parts : heme + globin Globin: four globin chains (2 α.>")


 Hb is found in RBCs its main function is to transport O2 to tissues. Structure: 2 parts : heme + globin Globin: four chains. Heme: porphyrin.>")



 Regionβ-Thalassaemiaα 0 -Thalassaemiaα + -Thalassaemia Americas 0–30–50–40.>")

