Download presentation
Presentation is loading. Please wait.

1
13 线粒体疾病 mitochondrial diseases

2
Mutations (changes) in the mitochondrial chromosome are responsible for a number of disorders.
 in the mitochondrial chromosome are responsible for a number of disorders.")
3
Mitochondrial disease is a chronic, genetic disorder that occurs when the mitochondria of the cell fails to produce enough energy for cell or organ function.

4
The incidence about 1:3000-4000 individuals in the US
The incidence about 1: individuals in the US. This is similar to the incidence of cystic fibrosis of caucasian births in the U.S.

5
There are many forms of mitochondrial disease
There are many forms of mitochondrial disease. Mitochondrial disease presents very differently from individual to individual.

6
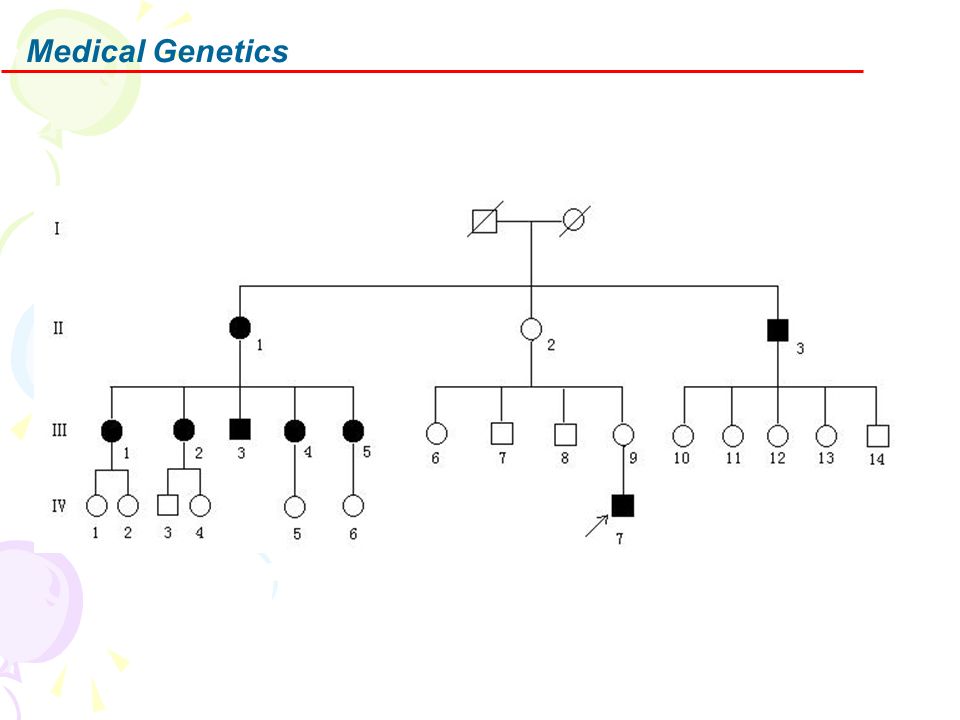
Mitochondrial disease is inherited in a number of different ways.
There may be one individual in a family or many individuals affected over a number of generations.

7
1. Biochemical & Genetic abnormalities of mitochondrial function
(1) Mitochondrial substrate transport (2) Substrate utilization (3) Citric acid (TCA) cycle (4) Oxidation-Phosphorylation coupling (5) Respiratory chain (6) Lipid membrane defect: (7) Nuclear gene defects
 Mitochondrial substrate transport (2) Substrate utilization (3) Citric acid (TCA) cycle (4) Oxidation-Phosphorylation coupling (5) Respiratory chain (6) Lipid membrane defect: (7) Nuclear gene defects.")
8
(1) Mitochondrial substrate transport
ATP/ADP translocator deficiency ATPase deficiency Carnitine-acylcarnitine translocase deficiency Carnitine deficiency Primary deficiency Secondary deficiency Carnitine palmitoyl transferase Protein import defects Solute carriers

9
(2) Substrate utilization
Pyruvate disorders β-oxidation defects (Fatty acid) Ketone synthesis HMG-CoA lyase HMG-CoA synthase
 Ketone synthesis HMG-CoA lyase HMG-CoA synthase.")
10
(3) Citric acid (TCA) cycle
Aconitase deficiency Lipoamide dehydrogenase Fumarase deficiency

11
(4) Oxidation-Phosphorylation coupling
ATP Synthase Luft's disease

12
(5) Respiratory chain mtDNA mutations Intergenomic communication Multiple mtDNA deletions mtDNA depletion Nuclear gene defects Tissue-specific Generalized Succinic dehydrogenase

13
(6) Lipid membrane defect
Barth

14
(7) Nuclear gene defects
 Nuclear gene defects")
15
2. The Symptoms of Mitochondrial Disease
Loss of muscle coordination, muscle weakness Neurological problems, seizures Visual and/or hearing problems Developmental delays, learning disabilities Heart, liver or kidney disease Gastrointestinal disorders, severe constipation Diabetes Increased risk of infection Thyroid and/or adrenal dysfunction Autonomic dysfunction Neuropsychological changes characterized by confusion, disorientation and memory loss.

16
3. LHON LHON = Leber's; Hereditary; Optic; Neuropathy

17
Genetic-Clinical correlations: Maternal Inheritance
Recurrence risks: Brother 30%; Sister 8%; Nephew 46%; Niece 10%; Male cousin 31%; Female cousin 6% 40% of patients with commonest mutation (G11778A) have negative family history Large families with maternal inheritance: G11778 & T14484C mutations
 have negative family history. Large families with maternal inheritance: G11778 & T14484C mutations.")
18
Mutations (General) 3 Mutations account for 96% of cases
All in Complex I genes Mutations: G11778A (69%), G3460A (13%), T14484C (14%) Mutation pattern Most patient's mutations are homoplasmic Some patients in each family may be heteroplasmic
, G3460A (13%), T14484C (14%) Mutation pattern. Most patient s mutations are homoplasmic. Some patients in each family may be heteroplasmic.")
19
Clinical features (General)
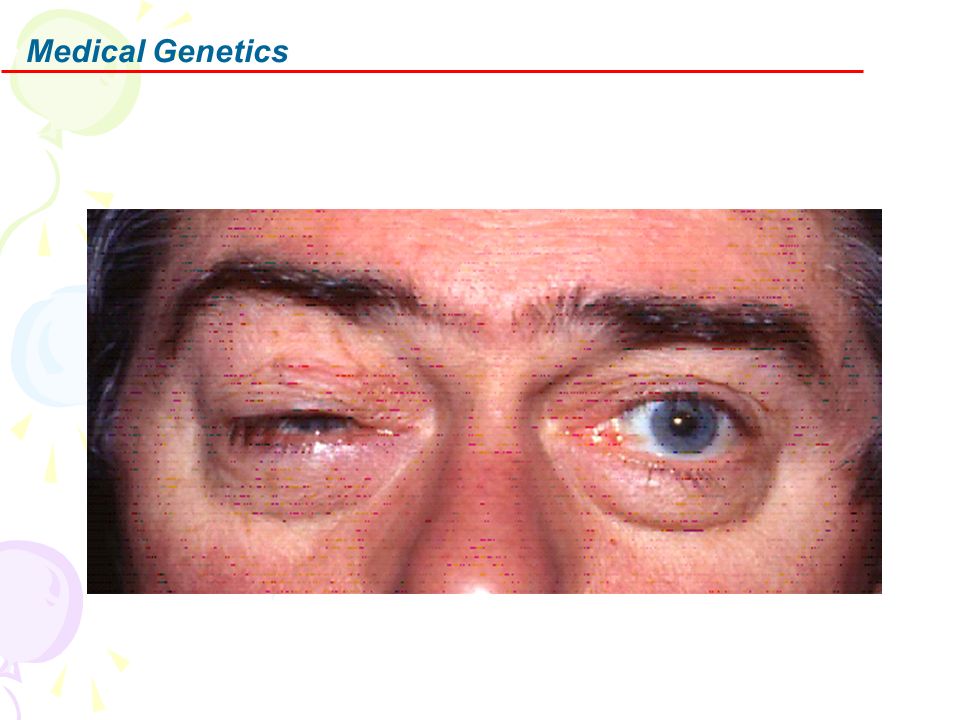
Male predominance:No relation to any X-linked genes Onset :Midlife: Mean 30 years; Range 1 to 70 Visual loss Clinical features Painless Visual loss pattern Severity: May deteriorate to 20/200 or less Progression: Mean 4 months; Interval between eyes affected: ~ 2 months Tendency to recover depends on mutation Pupillary reactions: May be relatively spared for degree of visual loss Ocular pathology Other features: Some families Cardiac conduction defects; Spastic dystonia; Spastic paraparesis; Dystonia

20
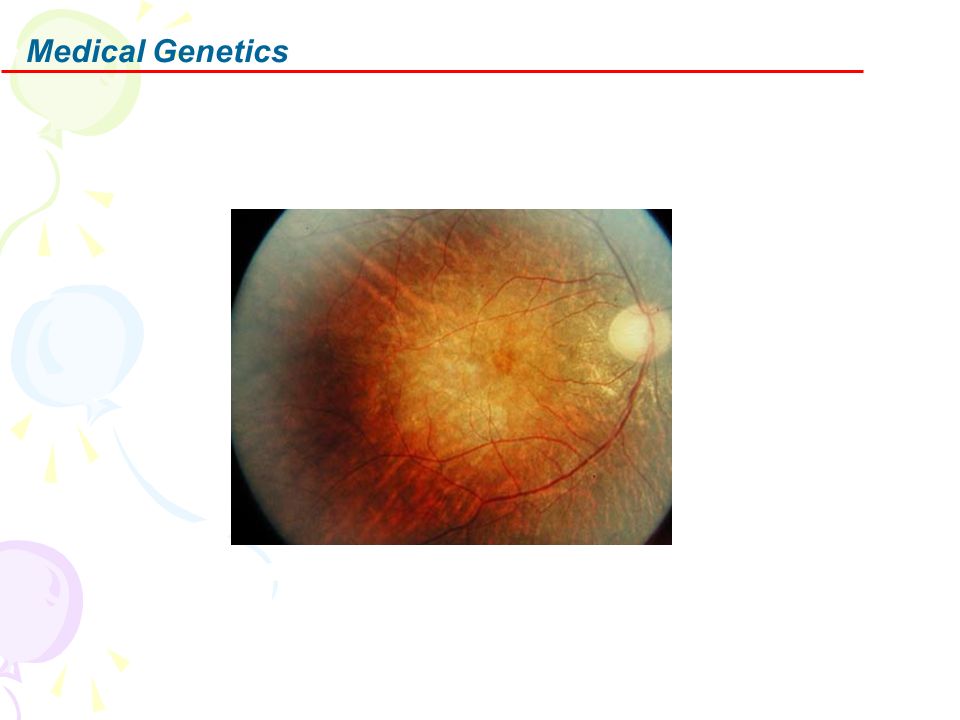
Bilateral, noncongruous central scotoma breaking through to upper periphery Associated with LHON

22
MRI: Optic nerve may enhance on T2 weighted images
Laboratory Muscle pathology No ragged red fibers EOM mitochondria: Diffuse increase in number and size; Disorganized cristae Preservation of myofibrils MRI: Optic nerve may enhance on T2 weighted images

23
4. MERRF MERRF=Myoclonic Epilepsy; Ragged Red Fibers

24
mtDNA point mutations: Heteroplasmic
tRNA Lys : A8344G (Frequent); T8356C; G8363A; G8361A Syndromes: MERRF or MERRF/MELAS overlap tRNA Ser Syndromes: MERRF/MELAS overlap; Epilepsia Partialis Continua; HAM tRNA Leu
; T8356C; G8363A; G8361A. Syndromes: MERRF or MERRF/MELAS overlap. tRNA Ser. Syndromes: MERRF/MELAS overlap; Epilepsia Partialis Continua; HAM. tRNA Leu.")
25
Onset Late adolescence - Early adult

26
Clinical syndrome: CNS Polyneuropathy (20%) Hearing loss (40%)
Myoclonus (60%) Epilepsy (45%) Cerebellar dysfunction: Ataxia Dementia Optic atrophy (20%) Polyneuropathy (20%) Distal sensory loss (large fiber modalities) Hearing loss (40%) Myopathy Short stature (10%) Lipomata (10%)
 Epilepsy (45%) Cerebellar dysfunction: Ataxia. Dementia. Optic atrophy (20%) Polyneuropathy (20%) Distal sensory loss (large fiber modalities) Hearing loss (40%) Myopathy. Short stature (10%) Lipomata (10%)")
28
Lactic acidosis: Variable Pathology of muscle
Laboratory Lactic acidosis: Variable Pathology of muscle Ragged red fibers: COX -

29
5. MELAS MELAS=Mitochondrial Encephalomyopathy; Lactic Acidosis; Stroke

30
Inheritance Maternal Occasional sporadic & non-inherited mutation (tRNA Leu) Clinical heterogeneity Rare to find more than 1 fully expressed MELAS in same family Maternal relatives often oligo- or asymptomatic

31
Heteroplasmic: Mutant mtDNA proportion ~ 56% to 95% Genes
mtDNA point mutations Heteroplasmic: Mutant mtDNA proportion ~ 56% to 95% Genes tRNA Leu (common) A3243G mutation: 80% of MELAS syndromes Other MELAS mutation loci: T3271C has later age of onset; 3291
 A3243G mutation: 80% of MELAS syndromes. Other MELAS mutation loci: T3271C has later age of onset;")
32
Encephalopathy: Often episodic Systemic features Myopathy
Clinical Syndrome Onset Mean = 10 years; Range = 2 to 40 Encephalopathy: Often episodic Systemic features Myopathy Polyneuropathy More common in patients with myopathy Mean life span with full clinical syndrome ~ 2 to 4 decades

33
Scattered "ragged red" muscle fibers: Gomori trichrome
Scattered abnormal, vacuolated fibers with clear rim: H & E

34
Ragged red muscle fibers: Gomori trichrome

35
Genetic counseling: A3423G mutation
% of affected offspring: Increased with higher mutant load in maternal blood Mutant load 1% to 19%: 20% chance of affected offspring Mutant load > 20%: 50% chance of affected offspring Full expression of phenotype in multiple family members: Rare Partial expression in multiple family members: Common

36
Laboratory Lactic acidosis: Blood & CSF EMG: Mormal or Myopathic
Serum CK: Normal to 2x high (32%) MRI: Strokes Biochemistry Respiratory chain dysfunction Reduced activity of Complexes I & IV Pathology (A3243G mutation)
 MRI: Strokes. Biochemistry. Respiratory chain dysfunction. Reduced activity of Complexes I & IV. Pathology (A3243G mutation)")
37
6. Kearns-Sayre Syndrome
Family history Sporadic: Most patients Familial cases: Rare; Mother to offspring

38
mtDNA mutation types Single large mtDNA deletion (2 to 8 kb)
Most common mutation type (80%) Common deletions Most common: 4977 base pairs from 8488 to 13460; 13 base pair repeat at mutation break point Thai patients: 3558 bp deletion; to 13761, or to 13765 Most deletions preserve Promoters of transcription of heavy & light strands 12S & 16S ribosomal RNA genes Origin of heavy strand replication Change in number of deletions over time Increased in muscle Reduced in rapidly turning over cells (hematopoetic) Large scale tandem duplication
 Common deletions. Most common: 4977 base pairs from 8488 to 13460; 13 base pair repeat at mutation break point. Thai patients: 3558 bp deletion; to 13761, or to Most deletions preserve. Promoters of transcription of heavy & light strands. 12S & 16S ribosomal RNA genes. Origin of heavy strand replication. Change in number of deletions over time. Increased in muscle. Reduced in rapidly turning over cells (hematopoetic) Large scale tandem duplication.")
39
Clinical features General
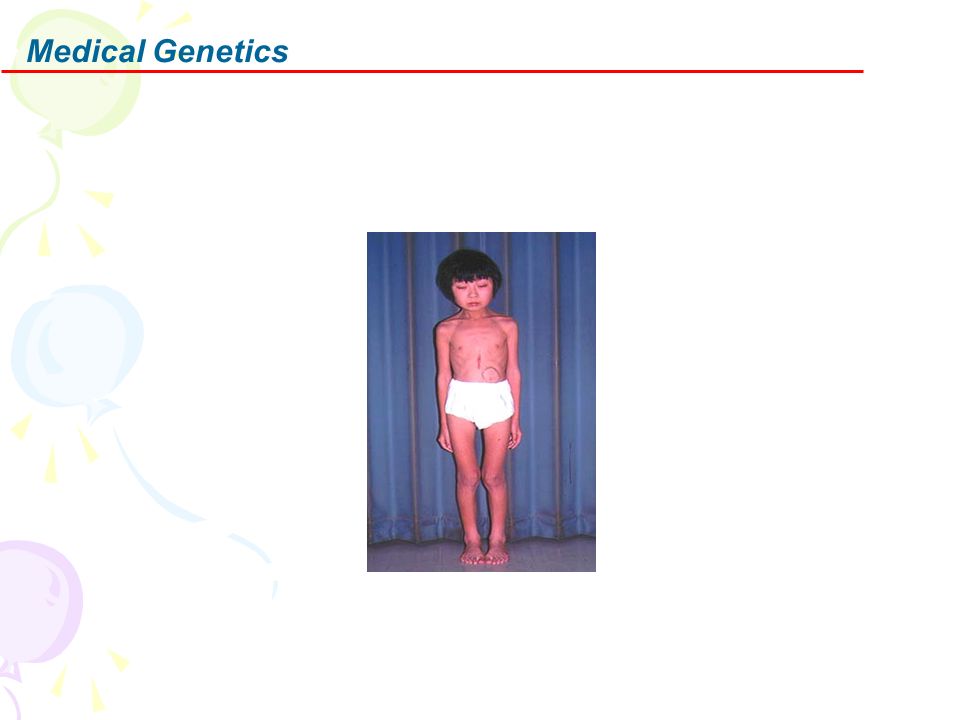
Characteristic signs: PEO; Pigmentary degeneration of retina; Heart block; Mitochondrial myopathy Onset: < 20 years; Later onset patients may have only PEO Ocular External Ophthalmoplegia Dysphagia: 50% of adults symptomatic Myopathy Weakness (90%): Proximal > Distal; Symmetric Occasional fatigue or pain on exertion CNS Hearing loss (95%) Ataxia (90%) Systemic features
: Proximal > Distal; Symmetric. Occasional fatigue or pain on exertion. CNS. Hearing loss (95%) Ataxia (90%) Systemic features.")
42
Laboratory Muscle pathology Lactic acidosis (80%)
Ragged red fibers (98%): COX + and COX - Variation in muscle fiber size Lactic acidosis (80%) Head CT: Basal ganglia calcifications (5%) CSF Protein: High
: COX + and COX - Variation in muscle fiber size. Lactic acidosis (80%) Head CT: Basal ganglia calcifications (5%) CSF Protein: High.")
43
6.Mitochondrial Disease Diagnosis
There is no reliable and consistent means of diagnosis. Diagnosis can be made by one of the few physicians that specializes in mitochondrial disease. Diagnosis can be made by blood DNA testing and/or muscle biopsy but neither of these tests are completely reliable.

44
7.Mitochondrial Disease Treatment
Treatment consists of vitamin therapy and conserving energy. The goal is to improve symptoms and slow progression of the disease. Conserve energy. Pace activities. Maintain an ambient environmental temperature. Avoid exposure to illness. Ensure adequate nutrition and hydration.

45
8. The Prognosis The prognosis is variable. Some people live a normal life and are minimally affected, others can be severely compromised with the disease. It is completely individualized The prognosis is unpredictable.

46
9. Future Goals To develop a better understanding of the biology of mitochondria. To learn more about mitochondrial disease in human beings. To refine diagnostic methods. To improve and expand treatment options. To educate the general public and medical arena. To improve the day to day life of individuals living with mitochondrial disease.

Similar presentations






 Dr. Mamoun Ahram Faculty of Medicine Second year, Second semester, 2014-2014 Principles.>")


>")








>")
