Download presentation
Presentation is loading. Please wait.

1
F Ahmadabadi F Ahmadabadi Child Neurologist ARUMS 20 14

2
Every child with unexplained... Neurological deterioration Metabolic acidosis Hypoglycemia Inappropriate ketosis Hypotonia Cardiomyopathy Hepatocellular dysfunction Failure to thrive... should be suspected of having a metabolic disorder

3
When to suspect an IEM? Clinical: Vomiting Lethargy FTT Seizure Respiratory Coma Cardiomyopathy Odor Abnormal hair Dysmorphology Labs: Metabolic acidosis Hypoglycemia Hyperammonemia Reducing substances in urine Ketonuria Pancytopenia

5
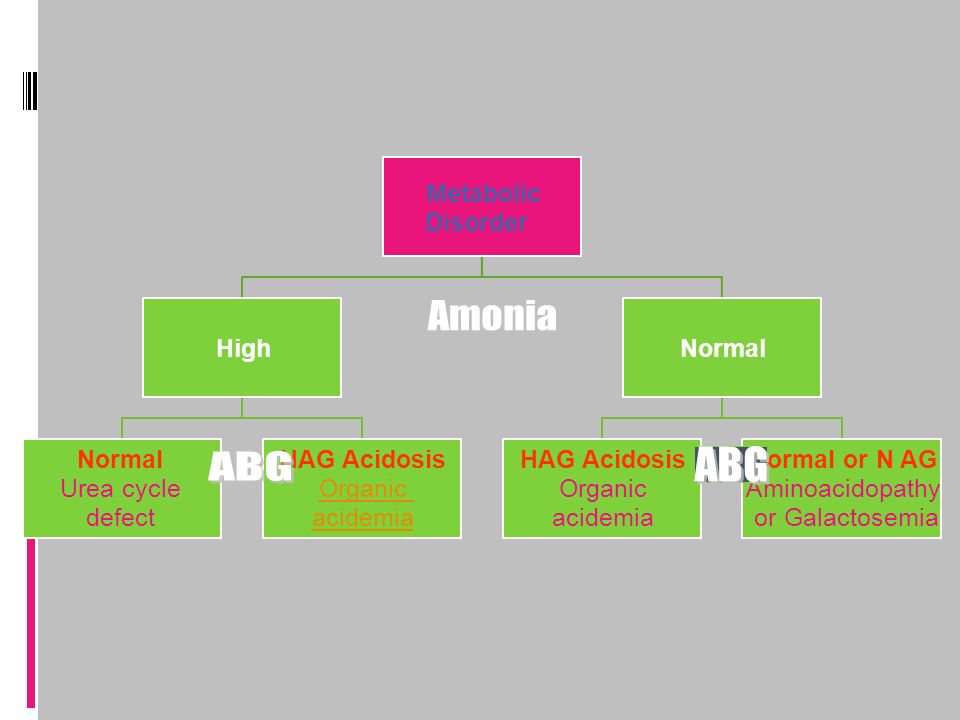
Organic Acidemia Ketosis No \ Skin manifestation MCD No Ketosis 1)Glutaric A 2)Acyl CoA deficiency 3)HMG CoA deficiency No skin manifestation No Odor 1)MMA 2)PPA Characteristic odor 1)MSUD 2)IVA
Glutaric A 2)Acyl CoA deficiency 3)HMG CoA deficiency No skin manifestation No Odor 1)MMA 2)PPA Characteristic odor 1)MSUD 2)IVA")
6
Screening 1)Gutherie 2)MS/MS Perform before discharge or 7 th day of birth ConfirmingAge of treatmentTestDisease Phenylalanin(p) AA (p)- AA (p) 1 st weeks of life Guthrie MS/MS PKU MSUD Thyrosinemia Organic acids(u) profile OAP (u)- AA (p) Biotinidase 1 st week of life 1 st weeks of life MS/MS Enzyme assessment Propionic acidemia Methyl mallonic Isovalleric Biotinidase deficiency GALT 1 st days of lifeEnzyme assessmentGalactosemia AA (p) profile DNA motations 1 st days of life MS/MS Urea cycle defect
Gutherie 2)MS/MS Perform before discharge or 7 th day of birth ConfirmingAge of treatmentTestDisease Phenylalanin(p) AA (p)- AA (p) 1 st weeks of life Guthrie MS/MS PKU MSUD Thyrosinemia Organic acids(u) profile OAP (u)- AA (p) Biotinidase 1 st week of life 1 st weeks of life MS/MS Enzyme assessment Propionic acidemia Methyl mallonic Isovalleric Biotinidase deficiency GALT 1 st days of lifeEnzyme assessmentGalactosemia AA (p) profile DNA motations 1 st days of life MS/MS Urea cycle defect")
7
Treatment in hyprammonemia 1.D/C oral intake temporarily 2.Usually IVF’s with glucose to give 12-15 mg/kg/min glu and at least 60 kcal/kg to prevent catabolism (may worsen PDH) 3.Bicarb/citrate 4.Carnitine/glycine 5.Na benzoate/arginine/citrulline 6.Dialysis--not exchange transfusion 7.Vitamins--often given in cocktails after labs drawn before dx is known Biotin, B6, B12, riboflavin, thiamine, folate
 3.Bicarb/citrate 4.Carnitine/glycine 5.Na benzoate/arginine/citrulline 6.Dialysis--not exchange transfusion 7.Vitamins--often given in cocktails after labs drawn before dx is known Biotin, B6, B12, riboflavin, thiamine, folate")
8
Metabolic Disorders Presenting as Severe Neonatal Disease 1. Disorders of Carbohydrate Metabolism Galactosemia - presents with severe liver disease, gram negative sepsis, and/or cataracts Enz deficiency: Gal-1-phos uridyl transferase, UDP-gal-4- epimerase Glycogen storage disease type 1a & 1b - presents as hypoglycemia Enz deficiency: Glucose-6 phosphatase Lactic Acidosis - presents as lactic acidosis +/- hypoglycemia Enz deficiency: Pyruvate carboxylase, Pyr dehydrogenase, etc. Fructose intolerance - Needs fructose exposure, hypoglycemia and acidosis

9
Metabolic Disorders Presenting as Severe Neonatal Disease 2. Amino Acid Disorders Maple syrup urine disease - presents with odor to urine and CNS problems Enz deficiency: Branched chain ketoacid decarboxylase Nonketotic hyperglycinemia - presents with CNS problems Enz deficiency: Glycine cleavage system Tyrosinemia - Severe liver disease, renal tubular dysfunction Enz deficiency: Fumaryl acetate Transient tyrosinemia of prematurity - progressive coma following respiratory distress

10
Metabolic Disorders Presenting as Severe Neonatal Disease 3. Urea Cycle Defects and Hyperammonemia 4. All present with lethargy, seizures, ketoacidosis, neutroenia, and hyperammonemia Ornithine carbamyl transferase (OTC) deficiency Carbamyl phosphate synthetase deficiency Citrullinemia Arginosuccinic Aciduria Argininemia Transient tyrosinemia of prematurity
 deficiency Carbamyl phosphate synthetase deficiency Citrullinemia Arginosuccinic Aciduria Argininemia Transient tyrosinemia of prematurity.")
11
Metabolic Disorders Presenting as Severe Neonatal Disease All present with lethargy, seizures, ketoacidosis, neutropenia, hyperammonemia, and/or hyperglycinemia 4. Organic Acid Defects Methylmalonic acidemia Proprionic acidemia Isovaleric acidemia - odor of “sweaty feet” Glutaric aciduria type II Dicarboxylic aciduria 5. Miscellaneous Peroxisomal disorders Lysosomal storage disease Pyridoxine dependent seizures

12
Amino acids metabolism diorders

13
Phenyl ketonuria AR Prevalence 1/10000 They are normal at birth.

14
Inheritance normal carrier GSD “Baby”

15
Phenylketonuria Phenylalanin Thyrosin Phenlketones

16
Clinical manifestation Normal at birth Severe MR(IQ<30) Blound appearance Odor Microcephalia Seborroic dermatitis Dominant maxilla
 Blound appearance Odor Microcephalia Seborroic dermatitis Dominant maxilla")
17
Diagnosis Ferric chloride test(urin phenyl ketones) Guthrie test Screening test MS/MS Phenylalanin>6mg/dlit(360 mic M) Diagnostic test Thyrosin (low)
 Guthrie test Screening test MS/MS Phenylalanin>6mg/dlit(360 mic M) Diagnostic test Thyrosin (low)")
18
Treatment Low phenylalanin Regimen (Phenylalanin2-6mg/dlit) Low phenylalanin Regimen Treatment must start in first 10 days of life. It must be continued till 10-12 yrs old. In malignant PKU, Neurotransmitters are needed.(BH4) Maternal hyperphenylallanenimia MR-Microcephallia- CHD +
 Maternal hyperphenylallanenimia MR-Microcephallia- CHD +.")
20
Carbohydrate metabolism disorders

21
1)Galactosemia Lactose Glucose+Galactose Galactose -1_phosphate Glucose-1-Phosphate
Galactosemia Lactose Glucose+Galactose Galactose -1_phosphate Glucose-1-Phosphate")
22
Clinical findings Feeding Hepatic failure (Bil –Coagulopathy -Glu) Tubulopathy (Acidosis-Glucosuria-A aciduria) Cataract E coli sepsis is more than others In older patients Learning disorders-Ovarian Failure Diagnosis: Screening Urin reducing substrare Diagnostic test RBCs Gal1-P U transferase
 Tubulopathy (Acidosis-Glucosuria-A aciduria) Cataract E coli sepsis is more than others In older patients Learning disorders-Ovarian Failure Diagnosis: Screening Urin reducing substrare Diagnostic test RBCs Gal1-P U transferase")
23
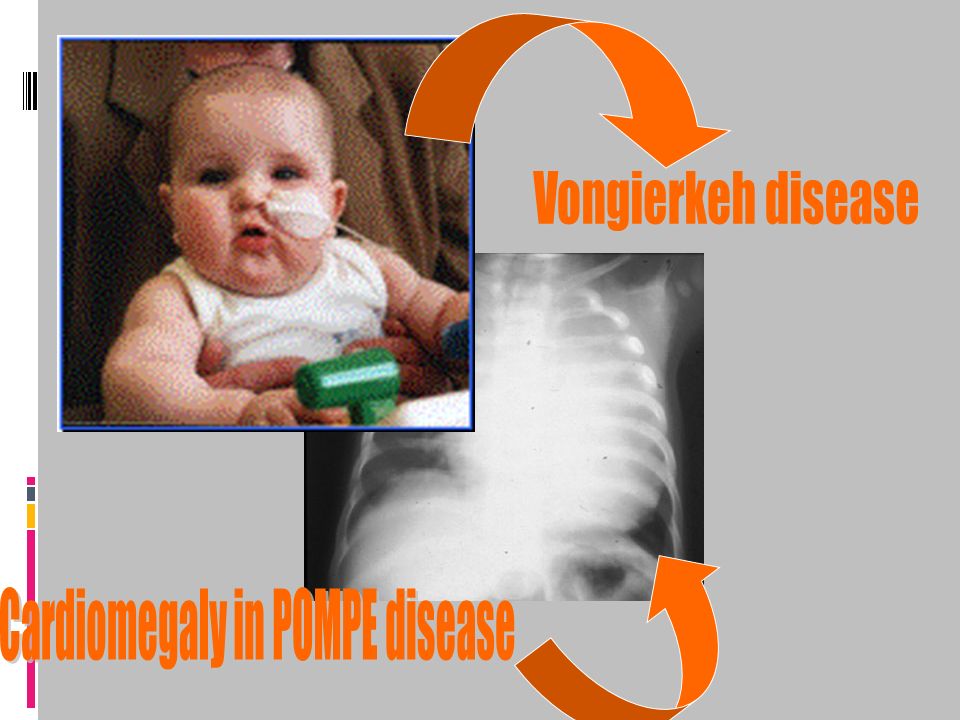
Type 0 Type I Type II Glycogen Storage Diseases Type IV Type VII

24
GSD Type III Type III

25
2)Glycogen storage diseases 1) liver involement &Hypoglycemia (1-6-8) 2) Muscle involvement (5-7) 3) Both of liver and Muscle (3) 4) Without any effect on Glucose & anearobic activities (2-4)
Glycogen storage diseases 1) liver involement &Hypoglycemia (1-6-8) 2) Muscle involvement (5-7) 3) Both of liver and Muscle (3) 4) Without any effect on Glucose & anearobic activities (2-4)")
27
Mucopolysaccharidosis B/MCNS involvement Organomega ly Retinal involvement Corneal involevment OnsetDisease Alder-reilly Severe ++_+1Yrs Hurler Alder-reilly Mild +++_1-2yrs Hunter Alder-reilly Severe + Liver __2-6 yrs Sanfilippo Alder-reilly Normal __+-2 yrs Morquo

28
Hurler syndrome

29
HURLER SYNDROME

31
LIPIDOSIS Onset Cherry red spot Organome galy 1 st month_+ Guacher disease ++ Nimenpic 3-6 month +_ Taysachs + Fabry 1 st 4 month + Farber

32
Treatment in hyprammonemia 1.D/C oral intake temporarily 2.Usually IVF’s with glucose to give 12-15 mg/kg/min glu and at least 60 kcal/kg to prevent catabolism (may worsen PDH) 3.Bicarb/citrate 4.Carnitine/glycine 5.Na benzoate/arginine/citrulline 6.Dialysis--not exchange transfusion 7.Vitamins--often given in cocktails after labs drawn before dx is known Biotin, B6, B12, riboflavin, thiamine, folate
 3.Bicarb/citrate 4.Carnitine/glycine 5.Na benzoate/arginine/citrulline 6.Dialysis--not exchange transfusion 7.Vitamins--often given in cocktails after labs drawn before dx is known Biotin, B6, B12, riboflavin, thiamine, folate")
Similar presentations















 Dr Mohammad Khassawneh Assistant professor of pediatrics.>")





