Download presentation
Presentation is loading. Please wait.

1
New methods for estimating species trees from genome-scale data Tandy Warnow The University of Illinois

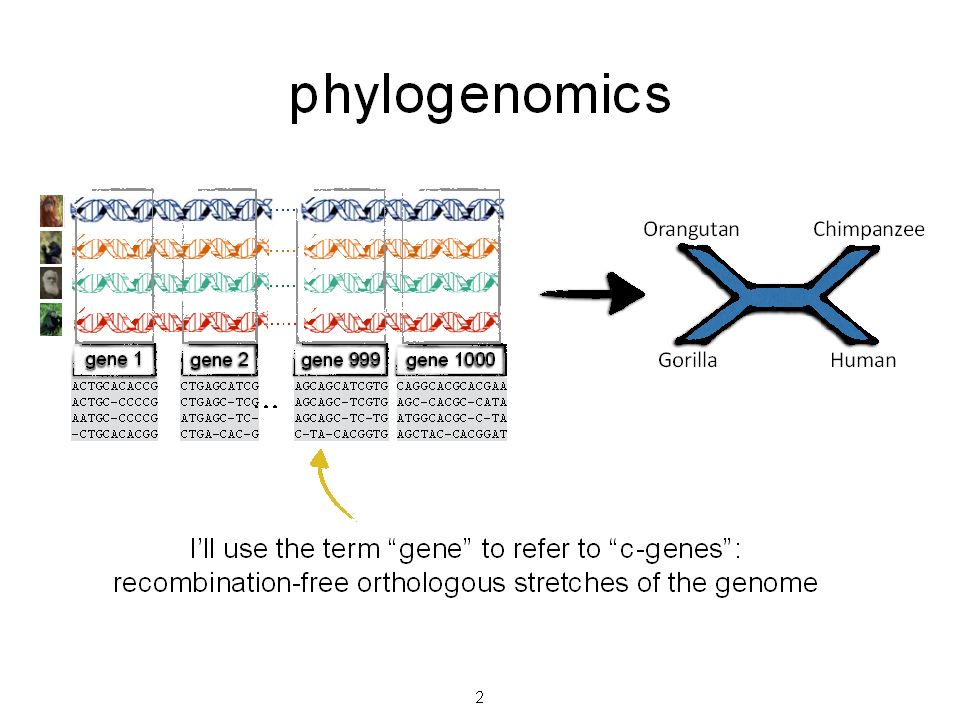
2
Orangutan GorillaChimpanzee Human From the Tree of the Life Website, University of Arizona Phylogeny (evolutionary tree)
")
3
Orangutan GorillaChimpanzee Human From the Tree of the Life Website, University of Arizona Sampling multiple genes from multiple species

5
Incomplete Lineage Sorting (ILS) is a dominant cause of gene tree heterogeneity
 is a dominant cause of gene tree heterogeneity")
6
This talk Gene tree heterogeneity due to incomplete lineage sorting, modelled by the multi-species coalescent (MSC) Statistically consistent estimation of species trees under the MSC, and the impact of gene tree estimation error “Statistical binning” (Science 2014) – improving gene tree estimation, and hence species tree estimation Open questions
 Statistically consistent estimation of species trees under the MSC, and the impact of gene tree estimation error Statistical binning (Science 2014) – improving gene tree estimation, and hence species tree estimation Open questions")
7
Gene trees inside the species tree (Coalescent Process) Present Past Courtesy James Degnan Gorilla and Orangutan are not siblings in the species tree, but they are in the gene tree.
 Present Past Courtesy James Degnan Gorilla and Orangutan are not siblings in the species tree, but they are in the gene tree.")
8
Incomplete Lineage Sorting (ILS) Confounds phylogenetic analysis for many groups: Hominids, Birds, Yeast, Animals, Toads, Fish, Fungi, etc. There is substantial debate about how to analyze phylogenomic datasets in the presence of ILS.

9
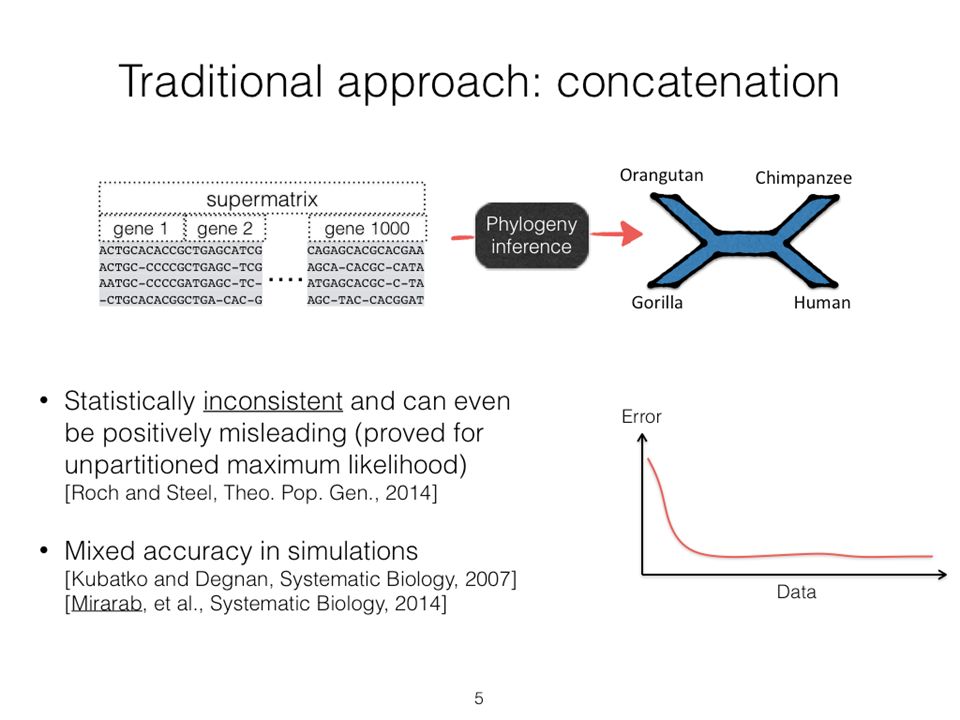
... Analyze separately Summary Method Two competing approaches gene 1 gene 2... gene k... Concatenation Species

10
Statistical Consistency error Data

12
... What about summary methods?

13
... What about summary methods? Techniques: Most frequent gene tree? Consensus of gene trees? Other?

14
Statistically consistent under ILS? Coalescent-based summary methods: – MP-EST (Liu et al. 2010): maximum pseudo-likelihood estimation of rooted species tree based on rooted triplet tree distribution – YES – BUCKy-pop (Ané and Larget 2010): quartet-based Bayesian species tree estimation –YES – And many others (ASTRAL, ASTRID, NJst, GLASS, etc.) Co-estimation methods: *BEAST (Heled and Drummond 2009): Bayesian co- estimation of gene trees and species trees – YES Single-site methods (SVDquartets, METAL, SNAPP, and others)
: maximum pseudo-likelihood estimation of rooted species tree based on rooted triplet tree distribution – YES – BUCKy-pop (Ané and Larget 2010): quartet-based Bayesian species tree estimation –YES – And many others (ASTRAL, ASTRID, NJst, GLASS, etc.) Co-estimation methods: *BEAST (Heled and Drummond 2009): Bayesian co- estimation of gene trees and species trees – YES Single-site methods (SVDquartets, METAL, SNAPP, and others).")
15
Statistically consistent under ILS? Coalescent-based summary methods: – MP-EST (Liu et al. 2010): maximum pseudo-likelihood estimation of rooted species tree based on rooted triplet tree distribution – YES – BUCKy-pop (Ané and Larget 2010): quartet-based Bayesian species tree estimation –YES – And many others (ASTRAL, ASTRID, NJst, GLASS, etc.) Co-estimation methods: *BEAST (Heled and Drummond 2009): Bayesian co- estimation of gene trees and species trees – YES Single-site methods (SVDquartets, METAL, SNAPP, and others) - YES CA-ML (Concatenation using unpartitioned maximum likelihood) - NO MDC – NO GC (Greedy Consensus) – NO MRP (supertree method) – NO
: maximum pseudo-likelihood estimation of rooted species tree based on rooted triplet tree distribution – YES – BUCKy-pop (Ané and Larget 2010): quartet-based Bayesian species tree estimation –YES – And many others (ASTRAL, ASTRID, NJst, GLASS, etc.) Co-estimation methods: *BEAST (Heled and Drummond 2009): Bayesian co- estimation of gene trees and species trees – YES Single-site methods (SVDquartets, METAL, SNAPP, and others) - YES CA-ML (Concatenation using unpartitioned maximum likelihood) - NO MDC – NO GC (Greedy Consensus) – NO MRP (supertree method) – NO.")
16
Statistically consistent under ILS? Coalescent-based summary methods: – MP-EST (Liu et al. 2010): maximum pseudo-likelihood estimation of rooted species tree based on rooted triplet tree distribution – YES – BUCKy-pop (Ané and Larget 2010): quartet-based Bayesian species tree estimation –YES – And many others (ASTRAL, ASTRID, NJst, GLASS, etc.) Co-estimation methods: *BEAST (Heled and Drummond 2009): Bayesian co- estimation of gene trees and species trees – YES Single-site methods (SVDquartets, METAL, SNAPP, and others) - YES CA-ML (Concatenation using unpartitioned maximum likelihood) - NO MDC – NO GC (Greedy Consensus) – NO MRP (supertree method) – NO
: maximum pseudo-likelihood estimation of rooted species tree based on rooted triplet tree distribution – YES – BUCKy-pop (Ané and Larget 2010): quartet-based Bayesian species tree estimation –YES – And many others (ASTRAL, ASTRID, NJst, GLASS, etc.) Co-estimation methods: *BEAST (Heled and Drummond 2009): Bayesian co- estimation of gene trees and species trees – YES Single-site methods (SVDquartets, METAL, SNAPP, and others) - YES CA-ML (Concatenation using unpartitioned maximum likelihood) - NO MDC – NO GC (Greedy Consensus) – NO MRP (supertree method) – NO.")
17
Results on 11-taxon datasets with weak ILS * BEAST more accurate than summary methods (MP-EST, BUCKy, etc) CA-ML (concatenated analysis) most accurate Datasets from Chung and Ané, 2011 Bayzid & Warnow, Bioinformatics 2013
 CA-ML (concatenated analysis) most accurate Datasets from Chung and Ané, 2011 Bayzid & Warnow, Bioinformatics 2013")
18
Problem: poor gene trees Summary methods combine estimated gene trees, not true gene trees. The individual gene sequence alignments in the 11-taxon datasets have poor phylogenetic signal, and result in poorly estimated gene trees. Species trees obtained by combining poorly estimated gene trees have poor accuracy.

19
Problem: poor gene trees Summary methods combine estimated gene trees, not true gene trees. The individual gene sequence alignments in the 11-taxon datasets have poor phylogenetic signal, and result in poorly estimated gene trees. Species trees obtained by combining poorly estimated gene trees have poor accuracy.

20
Problem: poor gene trees Summary methods combine estimated gene trees, not true gene trees. The individual gene sequence alignments in the 11-taxon datasets have poor phylogenetic signal, and result in poorly estimated gene trees. Species trees obtained by combining poorly estimated gene trees have poor accuracy.

21
Summary methods combine estimated gene trees, not true gene trees. The individual gene sequence alignments in the 11-taxon datasets have poor phylogenetic signal, and result in poorly estimated gene trees. Species trees obtained by combining poorly estimated gene trees have poor accuracy. TYPICAL PHYLOGENOMICS PROBLEM: many poor gene trees

22
Summary methods combine estimated gene trees, not true gene trees. The individual gene sequence alignments in the 11-taxon datasets have poor phylogenetic signal, and result in poorly estimated gene trees. Species trees obtained by combining poorly estimated gene trees have poor accuracy. THIS IS A KEY ISSUE IN THE DEBATE ABOUT HOW TO COMPUTE SPECIES TREES

23
Statistical Consistency for summary methods error Data Data are gene trees, presumed to be randomly sampled true gene trees.

24
Avian Phylogenomics Project E Jarvis, HHMI G Zhang, BGI Approx. 50 species, whole genomes, 14,000 loci Published Science 2014 MTP Gilbert, Copenhagen S. Mirarab Md. S. Bayzid, UT-Austin UT-Austin T. Warnow UT-Austin Plus many many other people… Challenges: Massive gene tree conflict suggestive of ILS Coalescent-based analysis using MP-EST produced tree that conflicted with concatenation analysis Most gene trees had very low bootstrap support, suggestive of gene tree estimation error

25
Avian Phylogenomics Project E Jarvis, HHMI G Zhang, BGI Approx. 50 species, whole genomes, 14,000 loci MTP Gilbert, Copenhagen S. Mirarab Md. S. Bayzid, UT-Austin UT-Austin T. Warnow UT-Austin Plus many many other people… Solution: Statistical Binning Improves coalescent-based species tree estimation by improving gene trees (Mirarab, Bayzid, Boussau, and Warnow, Science 2014) Avian species tree estimated using Statistical Binning with MP-EST (Jarvis, Mirarab, et al., Science 2014)
 Avian species tree estimated using Statistical Binning with MP-EST (Jarvis, Mirarab, et al., Science 2014).")
26
Gene Tree Estimation Error can be due to insufficient data error Data Data are sites in an alignment for a c-gene

27
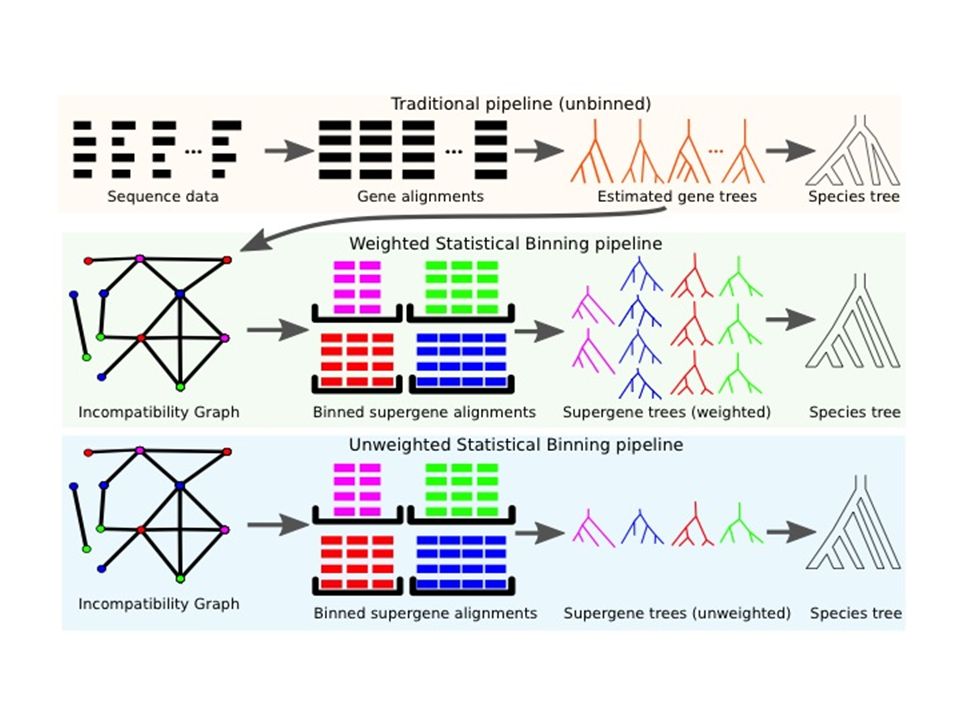
Unweighted statistical binning (Science 2014) Given multiple sequence alignments for a set of loci: 1.Estimate ML gene trees with bootstrap support 2.Bin genes based on gene tree compatibility after collapsing low support branches, producing “supergene alignments” 3.Compute “supergene trees” (one for each bin), using fully partitioned maximum likelihood 4.Apply coalescent-based summary method to the supergene trees, requiring that the summary method be statistically consistent under the MSC
 Given multiple sequence alignments for a set of loci: 1.Estimate ML gene trees with bootstrap support 2.Bin genes based on gene tree compatibility after collapsing low support branches, producing supergene alignments 3.Compute supergene trees (one for each bin), using fully partitioned maximum likelihood 4.Apply coalescent-based summary method to the supergene trees, requiring that the summary method be statistically consistent under the MSC")
28
Unweighted statistical binning pipelines are not statistically consistent under GTR+MSC Easy proof: As the number of sites per locus increase All estimated gene trees converge to the true gene tree and have bootstrap support that converges to 1 (Steel 2014) For every bin, with probability converging to 1, the genes in the bin have the same tree topology. Fully partitioned GTR ML analysis of each bin converges to a tree with the common topology of the genes in the bin. As the number of loci increase, every gene tree topology appears with probability converging to 1. Cannot infer the species tree from the flat distribution of gene trees!

29
Weighted statistical binning (PLOS One 2015) Given multiple sequence alignments for a set of loci: 1.Estimate ML gene trees with bootstrap support 2.Bin genes based on gene tree compatibility after collapsing low support branches, producing “supergene alignments” 3.Compute “supergene trees” (one for each bin), using fully partitioned maximum likelihood 4.Replace original gene tree by the new supergene tree (equivalently, replicate supergene trees by the size of each bin) 5.Apply coalescent-based summary method to the supergene trees, requiring that the summary method be statistically consistent under the MSC
 Given multiple sequence alignments for a set of loci: 1.Estimate ML gene trees with bootstrap support 2.Bin genes based on gene tree compatibility after collapsing low support branches, producing supergene alignments 3.Compute supergene trees (one for each bin), using fully partitioned maximum likelihood 4.Replace original gene tree by the new supergene tree (equivalently, replicate supergene trees by the size of each bin) 5.Apply coalescent-based summary method to the supergene trees, requiring that the summary method be statistically consistent under the MSC")
30
WSB pipelines are statistically consistent under GTR+MSC Easy proof: As the number of sites per locus increase All estimated gene trees converge to the true gene tree and have bootstrap support that converges to 1 (Steel 2014) For every bin, with probability converging to 1, the genes in the bin have the same tree topology Fully partitioned GTR ML analysis of each bin converges to a tree with the common topology of the genes in the bin Hence as the number of sites per locus and number of loci both increase, WSB followed by a statistically consistent summary method will converge in probability to the true species tree. Q.E.D.

32
Statistical binning vs. unbinned Mirarab, et al., Science 2014 (Unweighted statistical binning) Binning produces bins with approximate 5 to 7 genes each Datasets: 11-taxon strongILS datasets with 50 genes, Chung and Ané, Systematic Biology
 Binning produces bins with approximate 5 to 7 genes each Datasets: 11-taxon strongILS datasets with 50 genes, Chung and Ané, Systematic Biology.")
33
Comparing Binned and Un-binned MP-EST on the Avian Dataset Unbinned MP-EST strongly rejects Columbea, a major finding by Jarvis, Mirarab,et al. Binned MP-EST is largely consistent with the ML concatenation analysis. The trees presented in Science 2014 were the ML concatenation and Binned MP-EST

34
Summary Unpartitioned concatenation using maximum likelihood is statistically inconsistent under the MSC (Roch and Steel 2014, see discussion in Warnow PLOS Currents 2015) Gene tree estimation error impacts species tree estimation (multiple papers) Statistical binning (Mirarab et al. Science 2014) improves coalescent-based species tree estimation from multiple genes, used in Avian Tree (Jarvis, Mirarab, et al. Science 2014). Weighted statistical binning pipelines are statistically consistent under GTR+MSC, but unweighted statistical binning pipelines are not (Bayzid et al., PLOS One 2015)
 improves coalescent-based species tree estimation from multiple genes, used in Avian Tree (Jarvis, Mirarab, et al. Science 2014). Weighted statistical binning pipelines are statistically consistent under GTR+MSC, but unweighted statistical binning pipelines are not (Bayzid et al., PLOS One 2015).")
35
Bounded number of sites per locus? Do any summary methods converge to the species tree as the number of loci increase, but where each locus has only a constant number of sites? Roch & Warnow, Systematic Biology 2015: – Yes under the strong molecular clock (even for a single site per locus) – Very limited results otherwise
 – Very limited results otherwise.")
36
Open Questions Is fully partitioned ML statistically consistent or inconsistent under the MSC? (Note: proof by Roch and Steel for unpartitioned ML will not easily extend to fully partitioned) Are any of the standard summary methods statistically consistent for bounded number of sites per locus, but unbounded number of loci? Are the co-estimation methods (e.g., *BEAST and BEST) statistically consistent for bounded number of sites per locus but unbounded number of loci?
 Are any of the standard summary methods statistically consistent for bounded number of sites per locus, but unbounded number of loci. Are the co-estimation methods (e.g., *BEAST and BEST) statistically consistent for bounded number of sites per locus but unbounded number of loci .")
37
Open Questions Why does concatenation using ML (whether unpartitioned or partitioned) produce such good accuracy under many conditions? Why does statistical binning improve accuracy under many conditions?

38
Acknowledgments PhD students: Siavash Mirarab* (now Assistant Professor at UCSD ECE) and Md. S. Bayzid** Bastien Boussau (CNRS, Lyon) Sébastien Roch (Wisconsin) Funding: Guggenheim Foundation, Packard, NSF, Microsoft Research New England, David Bruton Jr. Centennial Professorship, TACC (Texas Advanced Computing Center), and GEBI. TACC and UTCS computational resources * Supported by HHMI Predoctoral Fellowship ** Supported by Fulbright Foundation Predoctoral Fellowship
 Sébastien Roch (Wisconsin) Funding: Guggenheim Foundation, Packard, NSF, Microsoft Research New England, David Bruton Jr. Centennial Professorship, TACC (Texas Advanced Computing Center), and GEBI. TACC and UTCS computational resources * Supported by HHMI Predoctoral Fellowship ** Supported by Fulbright Foundation Predoctoral Fellowship.")
Similar presentations



 Rebecca R. Gray, Ph.D. Department of Pathology University of Florida.>")















