Download presentation
Presentation is loading. Please wait.

1
Protein Structure Prediction: Homology Modeling & Threading/Fold Recognition D. Mohanty NII, New Delhi

3
Experimental Methods for Structure Determination

5
Computational Approaches for Protein Structure Prediction Methods based on laws of physical chemistry Ab initio folding using Molecular Mechanics Forcefield Knowledge-based Methods Homology Modelling Fold Recognition or Threading

7
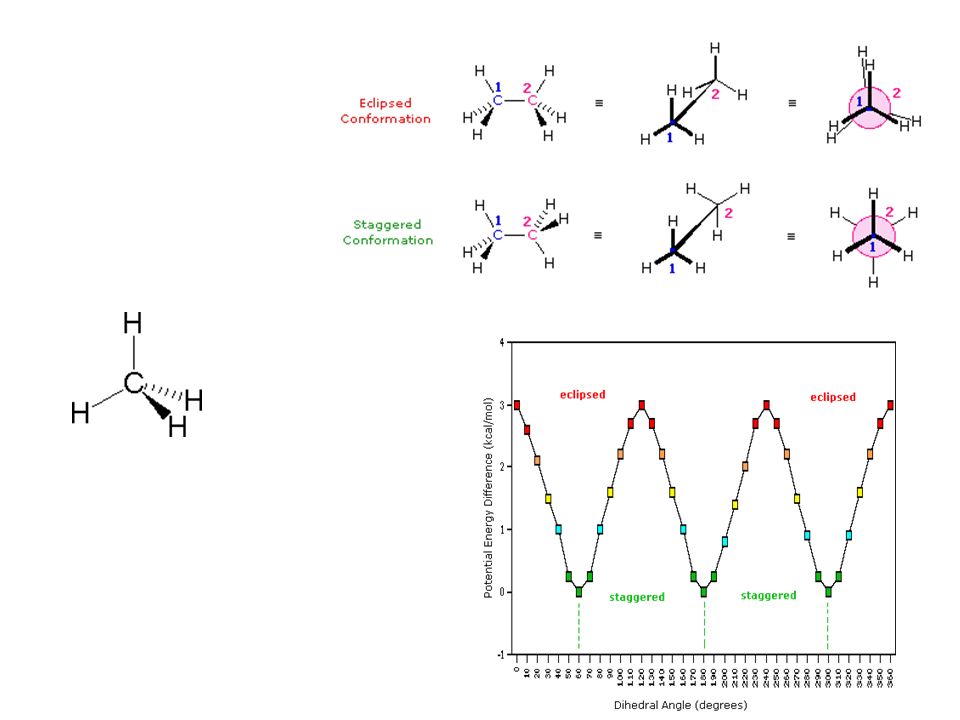
Interactions between atoms in a protein

10
Schematic depiction of the free energy surface of a protein Energy Minimization Molecular Dynamics Monte Carlo Simulations Computational tools for exploring energy surface & locating minimas

11
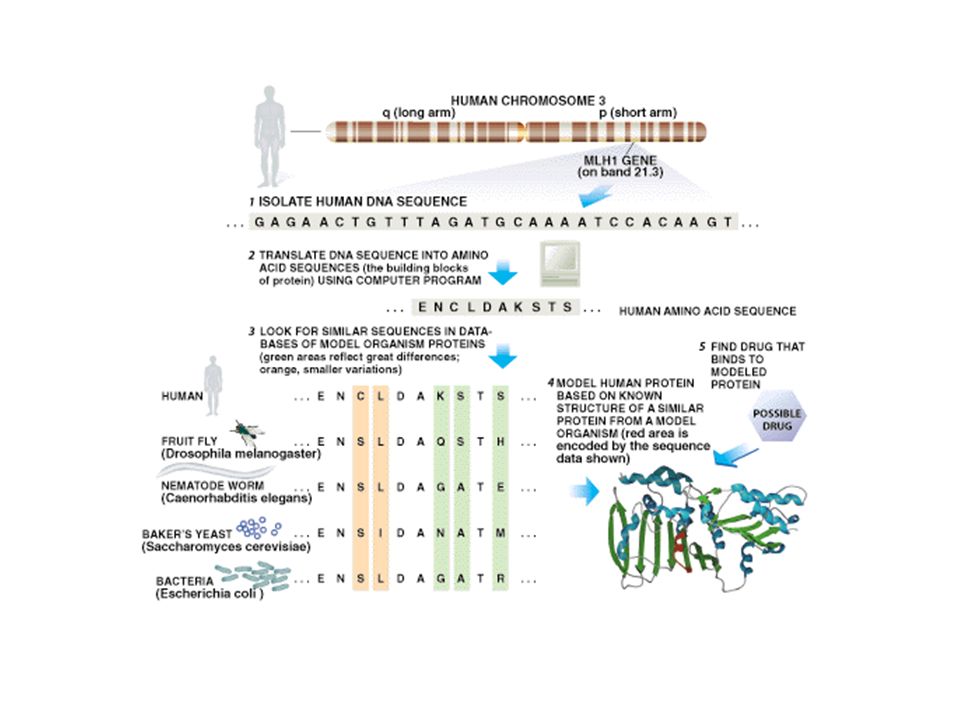
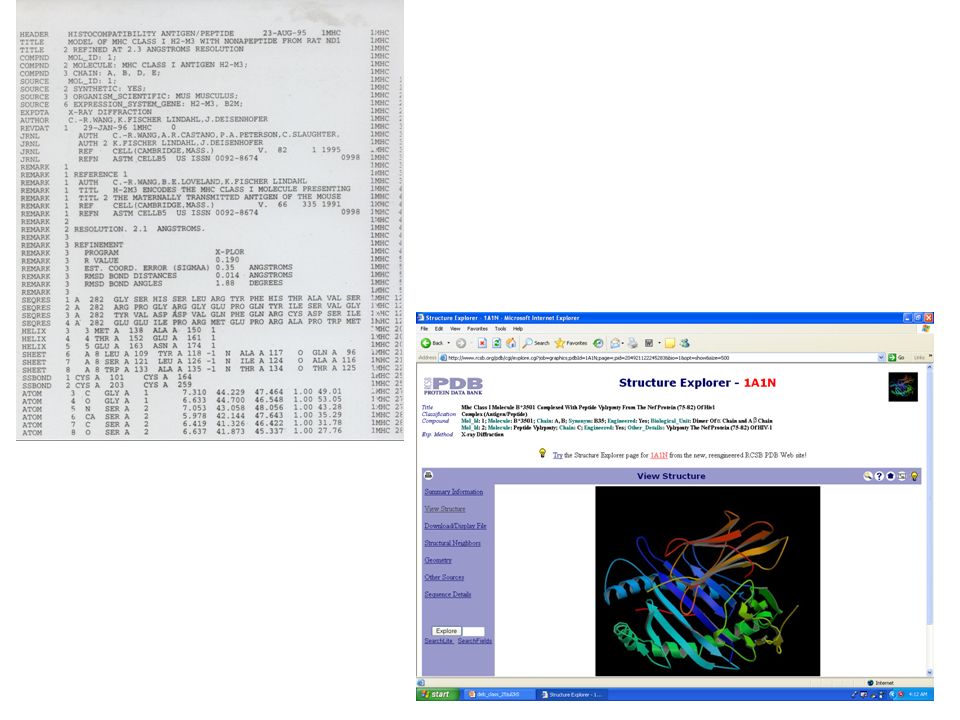
Structure Prediction Flowchart http://www.bmm.icnet.uk/people/rob/CCP11BBS/flowchart2.html

12
Homology Modelling Homology (or Comparative) modelling involves, building a 3D model for a protein of unknown structure (the target) on the basis of sequence similarity to proteins of known structure (the templates). Necessary requirements for homology modeling: Sequence similarity between the target and the template must be detectable. Substantially correct alignment between the target sequence and template must be calculated.

13
Homology or comparative modelling is Possible because: The 3D structures of the proteins in a family are more conserved than their sequences. Therefore, if similarity between two proteins is detectable at the sequence level, structural similarity can usually be assumed. Small changes in protein sequence usually results in small changes in 3D structure. But large changes in protein sequence can also result in small changes in its 3D structure i.e. Proteins with non-detectable sequence similarity can have similar structures.

14
Steps in Comparative Protein Structure Modelling

15
Target Template

16
Target Template

17
Simple sequence-sequence alignment using BLAST does not give alignment over the entire length.

18
Sidechain Modelling

19
Rotamer Library

20
Loop Modelling

21
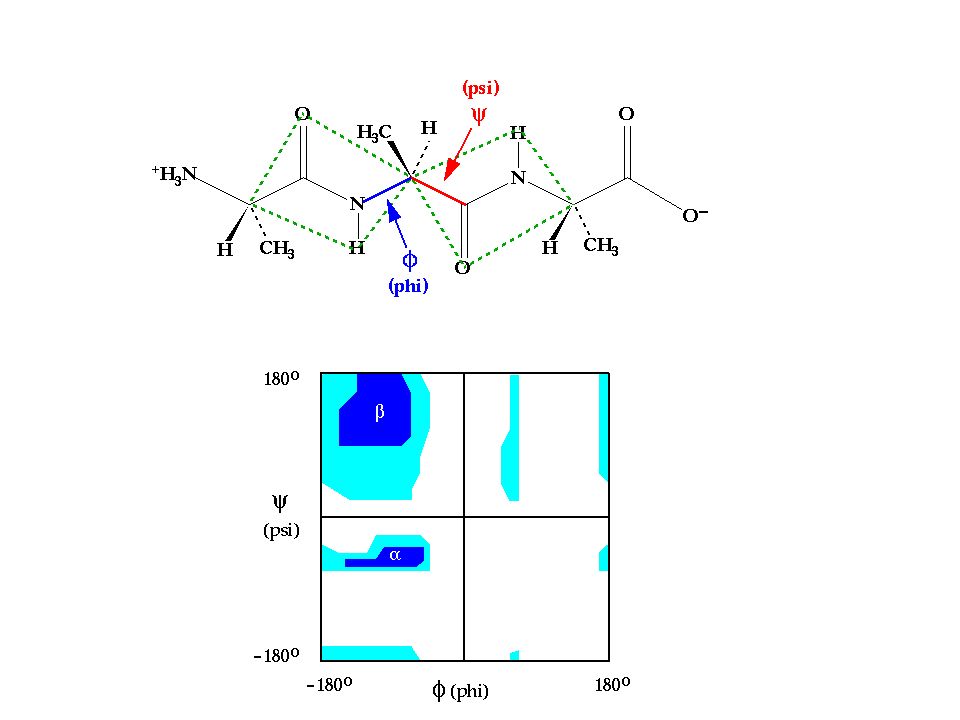
Model Validation Ramachandran Plot for backbone dihedrals Packing & Accessibility of amino acids

24
Threading or Fold Recognition Proteins often adopt similar folds despite no significant sequence or functional similarity. For many proteins there will be suitable template structures in PDB. Unfortunately, lack of sequence similarity will mean that many of these are undetected by sequence-only comparison done in homology modelling.

25
Goal of Fold Recognition or Threading Fold recognition methods attempt to detect the fold that is compatible with a particular query sequence. Unlike sequence-only comparison, these methods take advantage of the extra information made available by 3D structure. In effect, fold prediction methods turn the protein folding problem on its head: rather than predicting how a sequence will fold, they predict how well a fold will fit a sequence.

26
47% 17% 5%

29
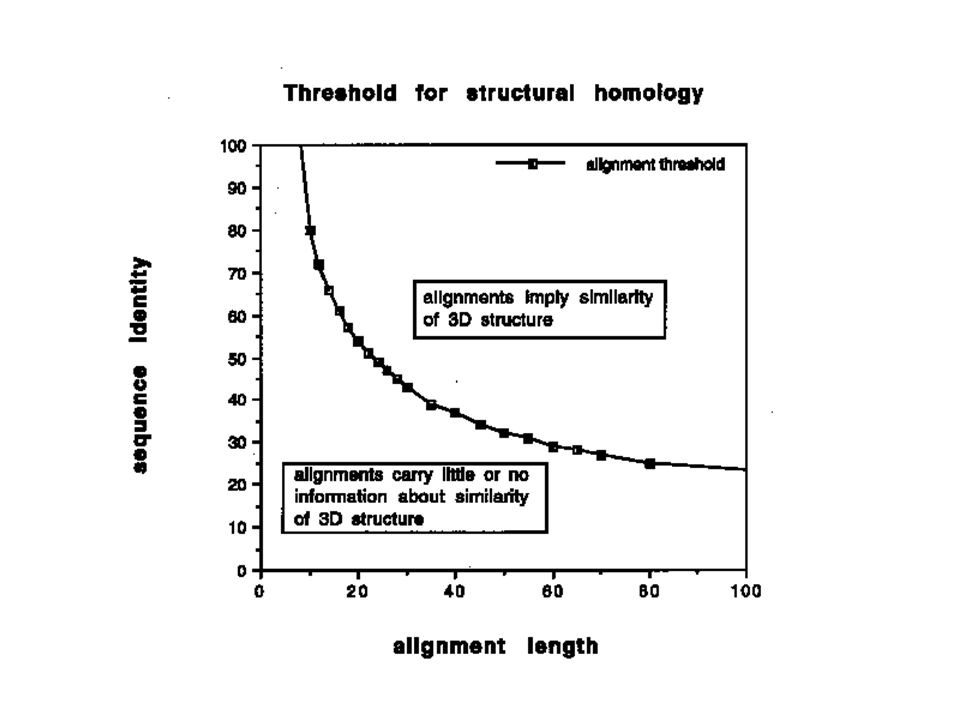
There are many examples of proteins exhibiting high structural similarity but less than 15% sequence identity. Classical sequence alignment fails to detect homology below 25-30% sequence identity. One needs sequence comparison methods which take into account structural environment of amino acids. Alternate approach is Threading or Fold Recognition, where sequence is compared directly to structure.

31
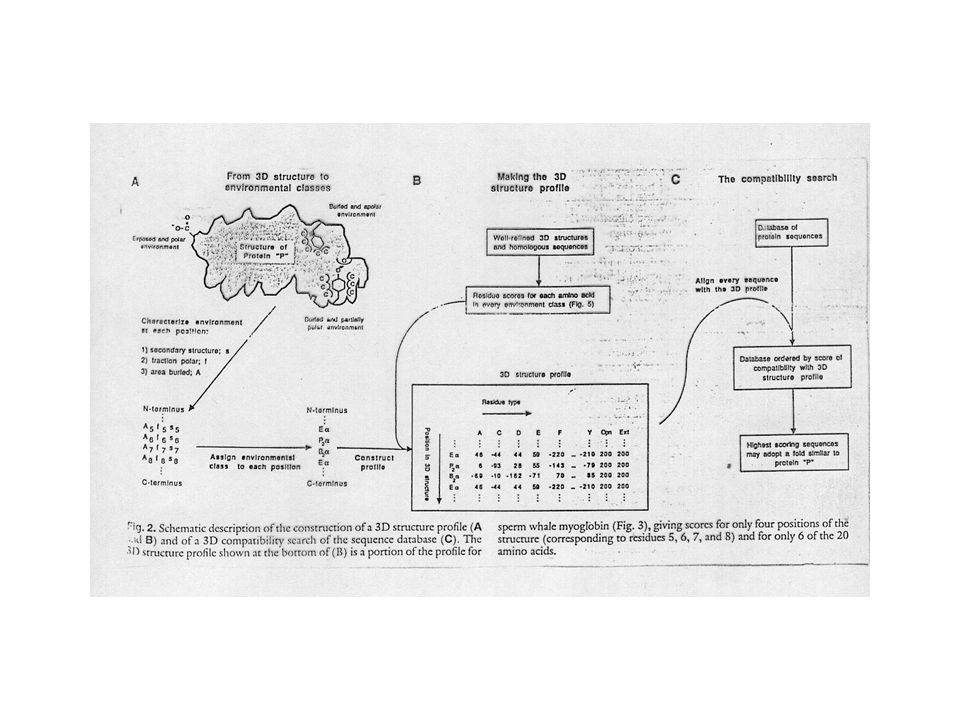
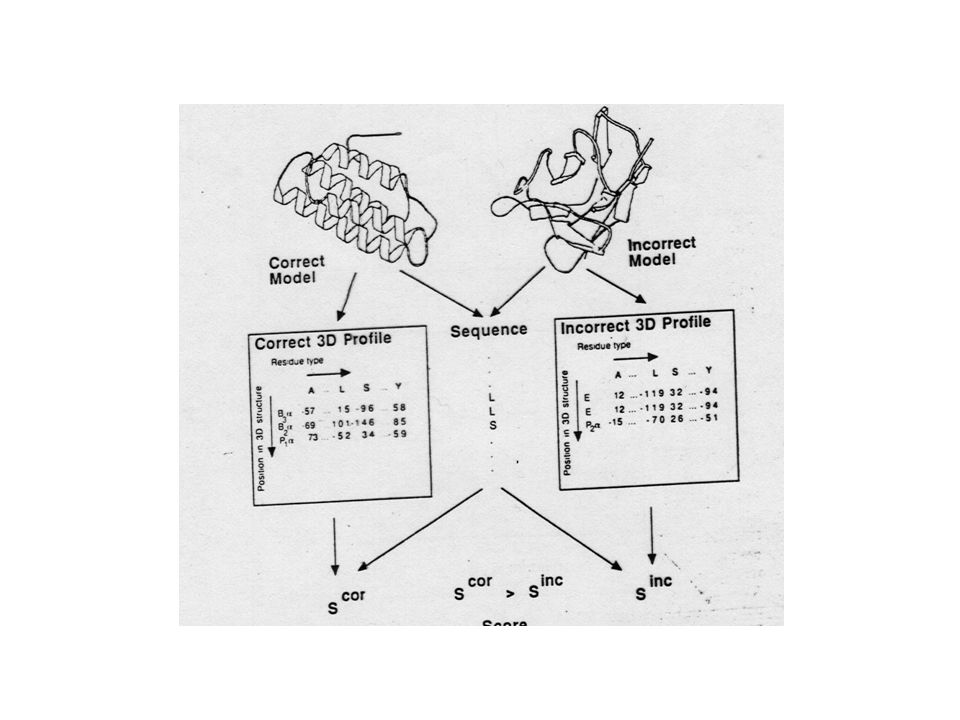
Compatibility of a sequence with a given fold

35
A practical approach for fold recognition Although fold prediction methods are not 100% accurate, the methods are still very useful. Run many different methods on many sequences from your homologous protein family. After all these runs, one can build up a consensus picture of the likely fold. Remember that a correct fold may not be at the top of the list, but it is likely to be in the top 10 scoring folds. Think about the function of your protein, and look into the function of the predicted folds. Don’t trust the alignments, rather use them as starting points.

36
Applications of comparative modeling. The potential uses of a comparative model depend on its accuracy. This in turn depends significantly on the sequence identity between the target and the template structure on which the model was based.

Similar presentations








![Structure Prediction. Tertiary protein structure: protein folding Three main approaches: [1] experimental determination (X-ray crystallography, NMR) [2]](/15/4859888/big_thumb.jpg "Structure Prediction. Tertiary protein structure: protein folding Three main approaches: [1] experimental determination (X-ray crystallography, NMR) [2]>")


.>")
![Structure Prediction. Tertiary protein structure: protein folding Three main approaches: [1] experimental determination (X-ray crystallography, NMR) [2]](/16/4993760/big_thumb.jpg "Structure Prediction. Tertiary protein structure: protein folding Three main approaches: [1] experimental determination (X-ray crystallography, NMR) [2]>")





>")
![. Protein Structure Prediction [Based on Structural Bioinformatics, section VII]](/16/5116302/big_thumb.jpg ". Protein Structure Prediction [Based on Structural Bioinformatics, section VII]>")



 Today: Why bother about proteins/prediction Concepts of molecular.>")