Download presentation
Presentation is loading. Please wait.

1
Introduction to Bioinformatics

2
Introduction to Bioinformatics.
LECTURE 6: Natural selection at the molecular basis * Chapter 6: Fighting HIV

3
6.1 Acquired Immune Deficiency Syndrome (AIDS)
Introduction to Bioinformatics LECTURE 6: NATURAL SELECTION AT THE MOLECULAR BASIS 6.1 Acquired Immune Deficiency Syndrome (AIDS) * First noticed in 1979 as peculiar disease in US * Only 1981 recognized as transmissible disease: AIDS * Infectious agent: HIV (Human immunodeficiency Virus) * Still not curable, more than 20 M victims, expensive medication (eg AZT) to keep the virus in check * How does HIV manage to evade our attempts to destroy it?
 * First noticed in 1979 as peculiar disease in US. * Only 1981 recognized as transmissible disease: AIDS. * Infectious agent: HIV (Human immunodeficiency Virus) * Still not curable, more than 20 M victims, expensive medication (eg AZT) to keep the virus in check. * How does HIV manage to evade our attempts to destroy it")
4
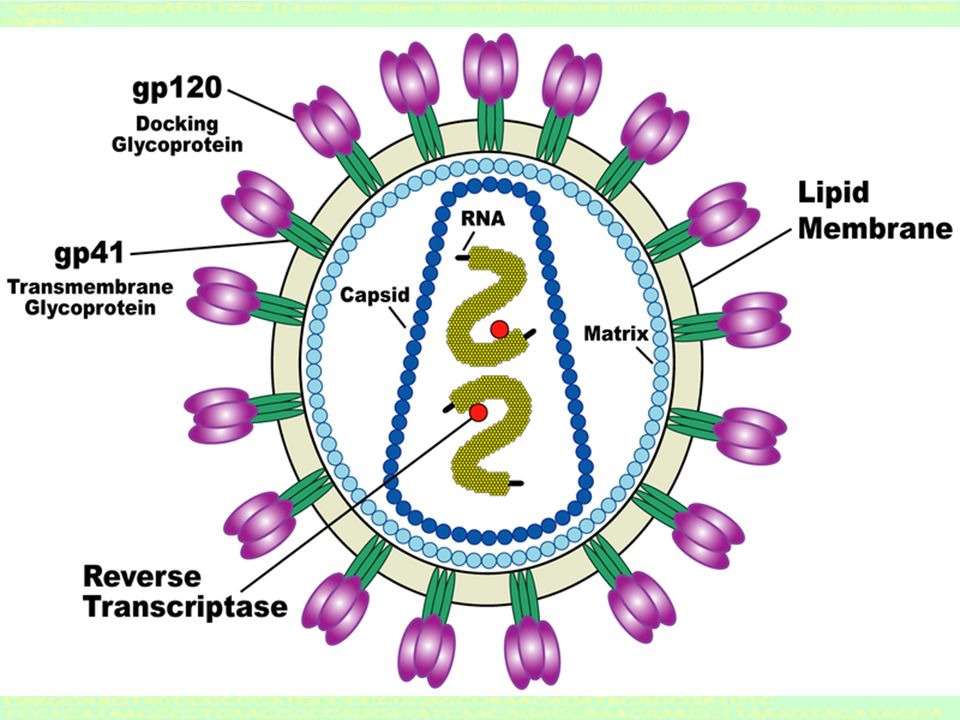
HIV virus

6
Introduction to Bioinformatics LECTURE 6: NATURAL SELECTION AT THE MOLECULAR BASIS
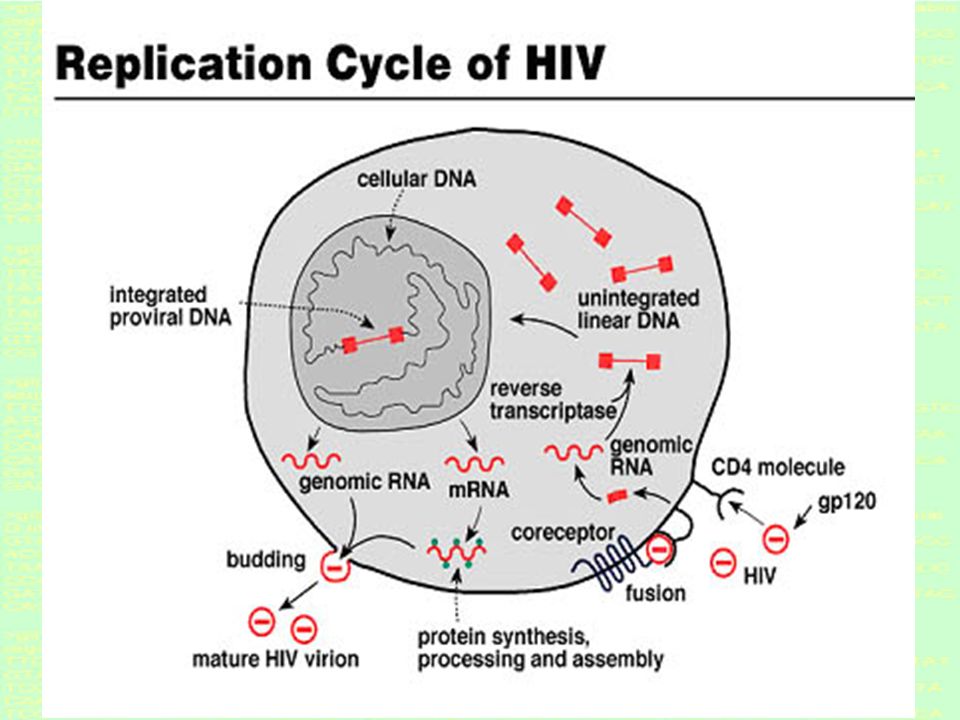
HIV is a retrovirus A retrovirus is an enveloped virus possessing a RNA genome, and replicate via a DNA intermediate. Retroviruses rely on the enzyme reverse-transcriptase to perform the reverse transcription of its genome from RNA into DNA, which can then be integrated into the host's genome with an integrase enzyme.

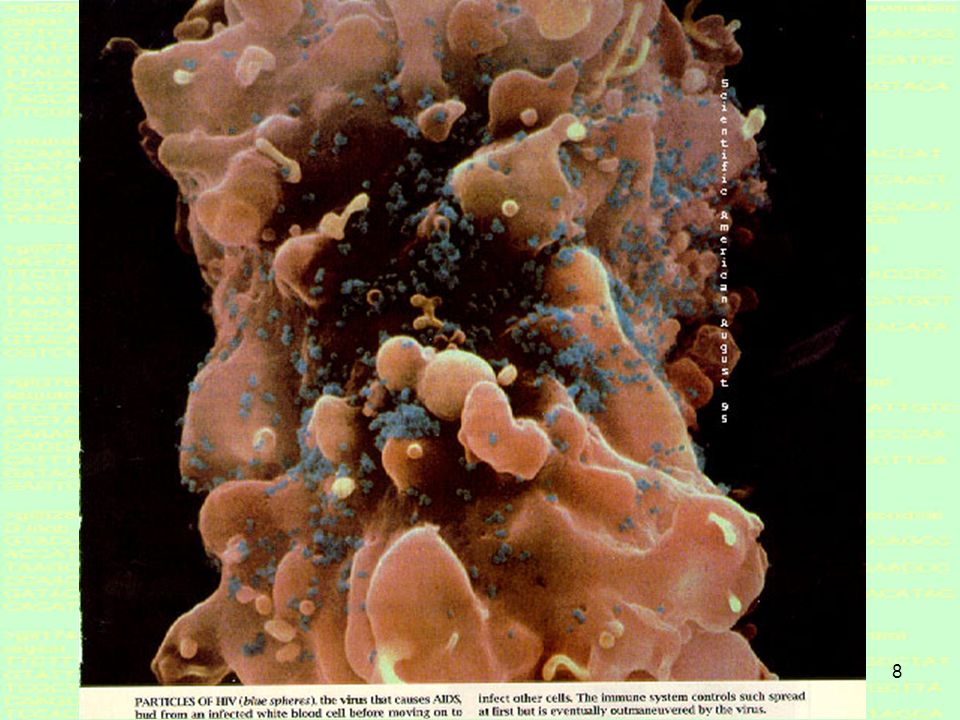
9
Scanning electron micrograph of HIV-1 budding from lymphocyte.

11
Introduction to Bioinformatics LECTURE 6: NATURAL SELECTION AT THE MOLECULAR BASIS

12
THE WORLD Mark Newman (

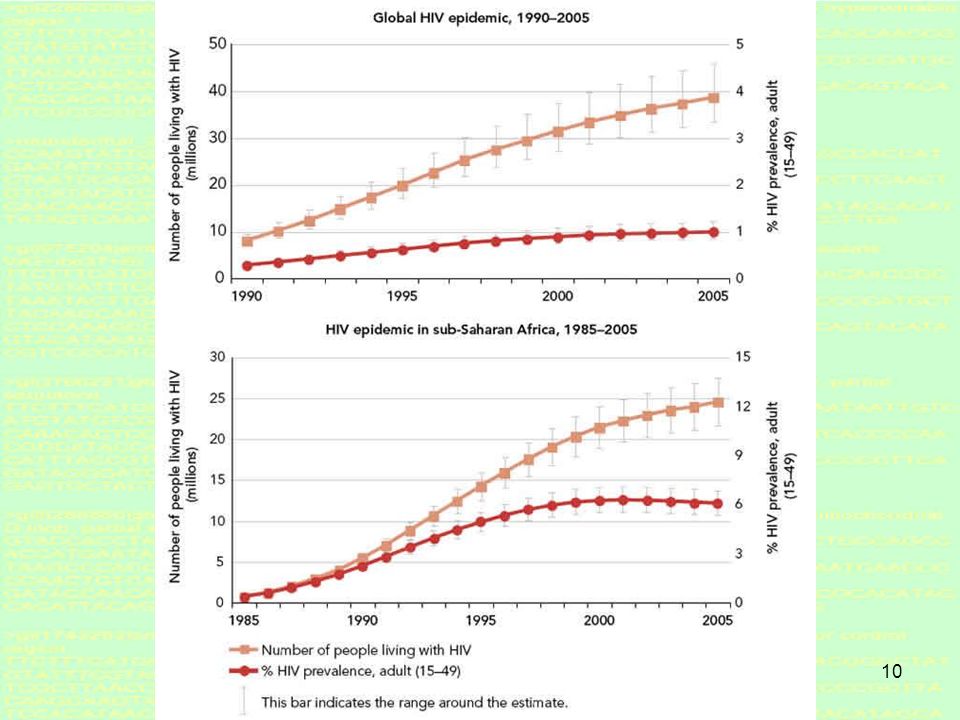
13
PEOPLE LIVING WITH HIV/AIDS
Mark Newman (

14
6.2 Evolution and natural selection
Introduction to Bioinformatics LECTURE 6: NATURAL SELECTION AT THE MOLECULAR BASIS 6.2 Evolution and natural selection 1859: Charles Darwin: on the origin of species by means of natural selection. At the molecular level: natural selection : * removes deleterious mutations: purifying or negative selection * Promotes spread of advantageous mutation: positive selection

15
6.3 HIV and the human immune system
Introduction to Bioinformatics LECTURE 6: NATURAL SELECTION AT THE MOLECULAR BASIS 6.3 HIV and the human immune system * HIV has a 9.5 Kb RNA genome - no DNA!!! * HIV is a retro-virus: RNA DNA virus * HIV recognizes helper T-cells of the human immune system * Infected T-cells have viral proteins sticking out that can be recognized by the immune system * Short reproduction span: 1.5 days to reproduce * RNA High error rate

16
Introduction to Bioinformatics 6.3: HIV and the human immune system
Fast reproduction + High error rate = FAST EVOLUTION Evolutionary arms race between human immune system and HIV

17
6.4 Quantifying natural selection on DNA sequences
Introduction to Bioinformatics LECTURE 6: NATURAL SELECTION AT THE MOLECULAR BASIS 6.4 Quantifying natural selection on DNA sequences * Mutations arise in the germ-line of one single individual and eventually become fixed in the population * We observe fixed mutations as differences between individuals * Most fixed mutations are neutral: genetic drift * Some 80-90% of the non-neutral mutations are detrimental to the organismal function. * A very small fraction of mutations is advantageous – but this is the engine for evolution.

18
* Synonymous and non-synonymous mutations.
Introduction to Bioinformatics 6.4 QUANTIFYING NATURAL SELECTION ON DNA SEQUENCES * How to measure whether mutations are neutral, deleterious, or advantageous? * Experimentally very difficult: short-lived simple organisms, and large populations (typical a virus) * Alternative: count number of mutations that can change the protein and those that don’t * Synonymous and non-synonymous mutations.
 * Alternative: count number of mutations that can change the protein and those that don’t. * Synonymous and non-synonymous mutations.")
19
Remember the translation from nucleotides to aminoacids
Introduction to Bioinformatics 6.4 QUANTIFYING NATURAL SELECTION ON DNA SEQUENCES Remember the translation from nucleotides to aminoacids (read from centre outwards)
")
20
* Non-synonymous mutations do not
Introduction to Bioinformatics 6.4 QUANTIFYING NATURAL SELECTION ON DNA SEQUENCES * Synonymous mutation: the new codon translates for the same amino-acid, example: GTT (Val) → GTA (Val). * Non-synonymous mutations do not * Mutations in the first position are sometimes synonymous (5%) * Mutations in the second position are never synonymous * Mutations in the third position are mostly synonymous
 → GTA (Val). * Non-synonymous mutations do not. * Mutations in the first position are sometimes synonymous (5%) * Mutations in the second position are never synonymous. * Mutations in the third position are mostly synonymous.")
21
* Almost all synonymous mutations are neutral.
Introduction to Bioinformatics 6.4 QUANTIFYING NATURAL SELECTION ON DNA SEQUENCES * Almost all synonymous mutations are neutral. * A priori, there are many more non-synonymous mutations possible than synonymous. * In most genes 70% of the mutations are non-synonymous * KA: #non-synonymous substitutions per non-synonymous site * KS: #synonymous substitutions per synonymous site

22
Introduction to Bioinformatics 6
Introduction to Bioinformatics 6.4 QUANTIFYING NATURAL SELECTION ON DNA SEQUENCES Motoo Kimura (1977): Comparison of the non-synonymous to the synonymous substitutions in a gene tells us about the strength and form of the natural selection, i.e.: the ratio KA / KS. Reasoning: * Advantageous mutations are very rare * Deleterious mutations will ‘not’ spread through a population * Therefore, most mutations are neutral Strong negative selection → Few non-synonymous substitutions
: Comparison of the non-synonymous to the synonymous substitutions in a gene tells us about the strength and form of the natural selection, i.e.: the ratio KA / KS. Reasoning: * Advantageous mutations are very rare. * Deleterious mutations will ‘not’ spread through a population. * Therefore, most mutations are neutral. Strong negative selection → Few non-synonymous substitutions.")
23
* f0 = fraction of non-synonymous mutations that are neutral.
Introduction to Bioinformatics 6.4 QUANTIFYING NATURAL SELECTION ON DNA SEQUENCES * f0 = fraction of non-synonymous mutations that are neutral. * v = mutation rate * # non-synonymous mutations after time t : KA = v f0 t * # synonymous mutations after time t : KS = v t * KA / KS = f0 * Strong negative selection: f0 is small thus KA / KS < 1 * If KA / KS is > 1 this is evidence for advantageous non-synonymous mutations

24
* Then after time t : KA = v(f0 + α)t * and: KA / KS = f0 + α
Introduction to Bioinformatics 6.4 QUANTIFYING NATURAL SELECTION ON DNA SEQUENCES * Define: α = fraction of non-synonymous mutations that are advantageous * Then after time t : KA = v(f0 + α)t * and: KA / KS = f0 + α * Thus KA / KS is gauge for the natural selection on genes * negative selection dominates: KA / KS < 1 * positive selection dominates: KA / KS > 1 * But averaged over the gene!
t. * and: KA / KS = f0 + α. * Thus KA / KS is gauge for the natural selection on genes. * negative selection dominates: KA / KS < 1. * positive selection dominates: KA / KS > 1. * But averaged over the gene!")
25
6.5 Estimating KA/KS How to determine KA/KS?
Introduction to Bioinformatics LECTURE 6: NATURAL SELECTION AT THE MOLECULAR BASIS 6.5 Estimating KA/KS How to determine KA/KS? Simplest way: just count and compare the number of synonymous and non-synonymous sites and ditto differences between two aligned strings Correct for multiple substitutions (e.g. Jukes-Cantor) Thus obtain a normalized ratio
 Thus obtain a normalized ratio.")
26
Introduction to Bioinformatics LECTURE 6: NATURAL SELECTION AT THE MOLECULAR BASIS
6.5 Estimating KA/KS Based upon this idea the algorithm of Masatoshi Nei and Takashi Gojobori (1986): Assume that rate of transitions and transversions is the same There is no bias towards codon usage (i.e. no information on the ensuing protein)
: Assume that rate of transitions and transversions is the same. There is no bias towards codon usage (i.e. no information on the ensuing protein)")
27
Introduction to Bioinformatics 6.5 ESTIMATING KA/KS
Nei-Gojobori algorithm * Consider two aligned homologous sequences without gaps s1 and s2 * Sc = #synonymous sites between s1 and s2 * Ac = #non-synonymous sites between s1 and s2 * Sd = #synonymous differences between s1 and s2 * Ad = #non-synonymous differences between s1 and s2

28
Introduction to Bioinformatics 6.5 ESTIMATING KA/KS
Nei-Gojobori algorithm * As the two sequences s1 and s2 are aligned there should be a correspondence between their codons. NOTE: point mutations only act on nucleotides and not on codons but here we analyse whether a mutation results in different aminoacids

29
Introduction to Bioinformatics 6.5 ESTIMATING KA/KS
Nei-Gojobori algorithm STEP 1: Count A and S sites

30
Introduction to Bioinformatics 6.5 NEI-GOJOBORI ALGORITHM
STEP 1: Count A and S sites Example: Consider the alignment : TTT TTA This is – say – the k-th codon of a sequence.

31
Introduction to Bioinformatics 6.5: NEI-GOJOBORI ALGORITHM
Now define: sc(ck) = #synonymous sites in this codon ac(ck) = 1 - sc(ck) = #non-synonymous sites in this codon fi : fraction of changes in at i-th position of codon that result in a synonymous change (i=1,2,3) Then: sc(ck) = ∑ fi and: ac(ck) = 3 - sc(ck) = 3 - ∑ fi
 = #synonymous sites in this codon. ac(ck) = 1 - sc(ck) = #non-synonymous sites in this codon. fi : fraction of changes in at i-th position of codon that result in a synonymous change (i=1,2,3) Then: sc(ck) = ∑ fi and: ac(ck) = 3 - sc(ck) = 3 - ∑ fi.")
32
Introduction to Bioinformatics 6.5: NEI-GOJOBORI ALGORITHM
In our example: Codon: TTA codes for: Leucine The 6 synonyms for Leucine (table 2.2 chapter 2, p. 27): CTA CTG CTC CTT TTA TTG f1 : 1 (ATA(-),GTA(-),CTA(+) from 3 changes, so: 1/3 f2 : 0 (TAA(-),TGA(-),TCA(-)) from 3 changes, so: 0/3 f3 : 1 (TTG(+),TTC(-),TTT(-)) from 3 changes, so: 1/3 So: sc(ck) = ∑ fi = 2/3 ac(ck) = 3 - sc(ck) = 3 - ∑ fi = 7/3
: CTA CTG CTC CTT TTA TTG. f1 : 1 (ATA(-),GTA(-),CTA(+) from 3 changes, so: 1/3. f2 : 0 (TAA(-),TGA(-),TCA(-)) from 3 changes, so: 0/3. f3 : 1 (TTG(+),TTC(-),TTT(-)) from 3 changes, so: 1/3. So: sc(ck) = ∑ fi = 2/3. ac(ck) = 3 - sc(ck) = 3 - ∑ fi = 7/3.")
33
Introduction to Bioinformatics 6.5: NEI-GOJOBORI ALGORITHM
For a DNA sequence of r codons: Sc = ∑k=1:r sc(ck) Ac = 3r - Sc For multiple sequences: average these quantities Note: do not include the STOP codon
 Ac = 3r - Sc. For multiple sequences: average these quantities. Note: do not include the STOP codon.")
34
Introduction to Bioinformatics 6.5: NEI-GOJOBORI ALGORITHM
STEP 2: Count A and S differences

35
Introduction to Bioinformatics 6.5: NEI-GOJOBORI ALGORITHM
Now define: sd(ck) = #synonymous differences in this codon ad(ck) = 1 - sd(ck) = #non-synonymous differences Example: sequence 1: GTT (Val) sequence 2: GTA (Val) there is only 1 difference and it is synonymous, so: sd = 1 and ad = 0
 = #synonymous differences in this codon. ad(ck) = 1 - sd(ck) = #non-synonymous differences. Example: sequence 1: GTT (Val) sequence 2: GTA (Val) there is only 1 difference and it is synonymous, so: sd = 1 and ad = 0.")
36
Introduction to Bioinformatics 6.5: NEI-GOJOBORI ALGORITHM
Multiple nucleotide differences between two codons: If there are n differences between two codons (n=0,1,2,3) then there are n! pathways from the first to the second codon Example: sequence 1: TTT (Phe) sequence 2: GTA (Val) the two possible pathways are : pathway 1 : TTT (Phe) ↔ GTT (Val) ↔ GTA (Val) pathway 2 : TTT (Phe) ↔ TTA (Leu) ↔ GTA (Val)
 then there are n! pathways from the first to the second codon. Example: sequence 1: TTT (Phe) sequence 2: GTA (Val) the two possible pathways are : pathway 1 : TTT (Phe) ↔ GTT (Val) ↔ GTA (Val) pathway 2 : TTT (Phe) ↔ TTA (Leu) ↔ GTA (Val)")
37
Introduction to Bioinformatics 6.5: NEI-GOJOBORI ALGORITHM
Example (Continued): the two possible pathways are : pathway 1 : TTT (Phe) ↔ GTT (Val) ↔ GTA (Val) pathway 2 : TTT (Phe) ↔ TTA (Leu) ↔ GTA (Val) Pathway 1 has: 1 non-syn and 1 syn substitution Pathway 2 has: 2 non-syn and 0 syn substitutions Assume that both pathways occur with same probability Therefore: sd = 1 syn / 2 pathways = 0.5 ad = 3 non-syns / 2 pathways = 1.5
: the two possible pathways are : pathway 1 : TTT (Phe) ↔ GTT (Val) ↔ GTA (Val) pathway 2 : TTT (Phe) ↔ TTA (Leu) ↔ GTA (Val) Pathway 1 has: 1 non-syn and 1 syn substitution. Pathway 2 has: 2 non-syn and 0 syn substitutions. Assume that both pathways occur with same probability. Therefore: sd = 1 syn / 2 pathways = 0.5. ad = 3 non-syns / 2 pathways = 1.5.")
38
Introduction to Bioinformatics 6.5: NEI-GOJOBORI ALGORITHM
For a codon with n differences: * Consider all n! pathways of n point-mutations * Evaluate sd and ad as above: * Average over all paths with equal weights * The total number of syn and non-syn differences is: Sd = ∑k=1:r sd(ck) Ad = ∑k=1:r ad(ck) Note: Sd + Ad is the total number of differences between the two sequences
 Ad = ∑k=1:r ad(ck) Note: Sd + Ad is the total number of differences between the two sequences.")
39
Introduction to Bioinformatics 6.5: NEI-GOJOBORI ALGORITHM
STEP 3: Compute KA and KS

40
Introduction to Bioinformatics 6.5: NEI-GOJOBORI ALGORITHM
* Approximate the proportion of synonymous (ds) and non-synonymous differences by: and * Use the Jukes-Cantor correction to find the number of substitutions: For both ds and da to obtain KS and KA.
 and non-synonymous differences by: and. * Use the Jukes-Cantor correction to find the number of substitutions: For both ds and da to obtain KS and KA.")
41
Introduction to Bioinformatics 6.5: NEI-GOJOBORI ALGORITHM
SUMMARY of Nei-Gojobori algorithm: see box on page 105 of the book Remark: the algorithm is linear in the size of the sequences

42
6.6 Case study: natural selection and the HIV genome
Introduction to Bioinformatics LECTURE 6: NATURAL SELECTION AT THE MOLECULAR BASIS 6.6 Case study: natural selection and the HIV genome * HIV is a fast evolving virus * HIV is a different kind of virus and has RNA and no DNA * An analysis of KA/KS over a gene is not so informative as it averages over positive and negative selection * Sliding window plot gives information on smaller scale of evolution pressure.

43
Introduction to Bioinformatics LECTURE 6: NATURAL SELECTION AT THE MOLECULAR BASIS

44
6.6 Case study: natural selection and the HIV genome
Introduction to Bioinformatics LECTURE 6: NATURAL SELECTION AT THE MOLECULAR BASIS 6.6 Case study: natural selection and the HIV genome * STEP 1: ORF finding

45
Introduction to Bioinformatics LECTURE 6: NATURAL SELECTION AT THE MOLECULAR BASIS
HIV-I genome

46
6.6 Case study: natural selection and the HIV genome
Introduction to Bioinformatics LECTURE 6: NATURAL SELECTION AT THE MOLECULAR BASIS 6.6 Case study: natural selection and the HIV genome * STEP 1: ORF finding * STEP 2: Nei-Gojobori to find high KA/KS ratios with sliding window plot.

47
HIV epitopes: the ENV gene
Introduction to Bioinformatics LECTURE 6: NATURAL SELECTION AT THE MOLECULAR BASIS HIV epitopes: the ENV gene An epitope is the part of a macromolecule that is recognized by the immune system, specifically by antibodies. ENV: Envelope and docking: strong selection pressure from human immune system

48
Introduction to Bioinformatics LECTURE 6: NATURAL SELECTION AT THE MOLECULAR BASIS

49
HIV epitopes: the GAG polyprotein
Introduction to Bioinformatics LECTURE 6: NATURAL SELECTION AT THE MOLECULAR BASIS HIV epitopes: the GAG polyprotein 1500 bp : viral core Strong selection pressure from human immune system

50
Introduction to Bioinformatics LECTURE 6: NATURAL SELECTION AT THE MOLECULAR BASIS

51
Introduction to Bioinformatics LECTURE 6: NATURAL SELECTION AT THE MOLECULAR BASIS
Visualisation of the fast evolution of the HIV virus with a phylogenetic tree

52
END of LECTURE 6

Similar presentations






 A Design by Jeanine Nasser.>")




Negative.>")



 confers some increase in the fitness of the organism Selection acts to favour this allele Also called adaptive.>")


>")
