Download presentation
Presentation is loading. Please wait.

1
Bioinformatics 生物信息学理论和实践 唐继军 jtang@cse.sc.edu 北京林业大学计算生物学中心 www.bjfuccb.com

2
Download and install programs Unzip or untar unzip If file.tar.gz, tar xvfz file.tar.gz Go to the directory and “./configure” Then “make”

3
System subroutine system ("ls –ltr");
;")
4
sub ReadFasta { my ($fname) = @_; open(FILE, $fname) or die "Cannot open $fname\n"; my $data = ""; my @dnas = (); while(my $line = ) { if ($line =~ /^>/) { if ($data ne "") { push(@dnas, $data); } $data = ""; } $data.= $line; } if ($data ne "") { push(@dnas, $data); } close FILE; return @dnas; }
 open(FILE, $fname) or die Cannot open $fname\n ; my $data = ; = (); while(my $line = ) { if ($line =~ /^>/) { if ($data ne ) { $data); } $data = ; } $data.= $line; } if ($data ne ) { $data); } close FILE; }")
5
print "Please input file name:\n"; my $fname = ; my @dnas = ReadFasta($fname); my $len = $#dnas + 1; for (my $i = 0; $i < $len; $i++) { for (my $j = $i+1; $j < $len; $j++) { for (my $k = $j+1; $k < $len; $k++) { $fname = "$i\_$j\_$k"; print $fname; open(OUT, ">$fname"); print OUT $dnas[$i]; print OUT $dnas[$j]; print OUT $dnas[$k]; close OUT; system ("./clustalw2 $i\_$j\_$k"); }
![print Please input file name:\n ; my $fname = ; = ReadFasta($fname); my $len = $#dnas + 1; for (my $i = 0; $i < $len; $i++) { for (my $j = $i+1; $j < $len; $j++) { for (my $k = $j+1; $k < $len; $k++) { $fname = $i\_$j\_$k ; print $fname; open(OUT, >$fname ); print OUT $dnas[$i]; print OUT $dnas[$j]; print OUT $dnas[$k]; close OUT; system ( ./clustalw2 $i\_$j\_$k ); }](http://images.slideplayer.com/24/6964946/slides/slide_5.jpg "print Please input file name:\n ; my $fname = ; = ReadFasta($fname); my $len = $#dnas + 1; for (my $i = 0; $i < $len; $i++) { for (my $j = $i+1; $j < $len; $j++) { for (my $k = $j+1; $k < $len; $k++) { $fname = $i\_$j\_$k ; print $fname; open(OUT, >$fname ); print OUT $dnas[$i]; print OUT $dnas[$j]; print OUT $dnas[$k]; close OUT; system ( ./clustalw2 $i\_$j\_$k ); }")
6
Debug Notice there are problems in a program is hard Find the source of the problem is even harder Good debug tool: print Better tool: debugger

7
Perl debugger perl –d program arguments n: next line s: step in r: run until the end of the current sub, repeat c: continue to the next breakpoint

8
Check source l List next several lines l 8-10 List line 8-10 l 100 List line 100 l subname List subroutine subname f restrcit.pl Switch to view restrict.pl

9
Breakpoint b 100 Add a breakpoint at line 100 of the current file b subname Add a breakpoint at this subroutine B Remove a break point B 100 will remove a breakpoint at line 100 B * will remove all breakpoints

10
See variable p $var Print the value of the variable y var Display my variable V display variables V var w $var Watch this var, stop when the value is changed

11
Working with Single DNA Sequences

12
Learning Objectives Discover how to manipulate your DNA sequence on a computer, analyze its composition, predict its restriction map, and amplify it with PCR Find out about gene-prediction methods, their potential, and their limitations Understand how genomes and sequences and assembled

13
Outline 1.Cleaning your DNA of contaminants 2.Digesting your DNA in the computer 3.Finding protein-coding genes in your DNA sequence 4.Assembling a genome

14
Cleaning DNA Sequences In order to sequence genomes, DNA sequences are often cloned in a vector (plasmid, YAC, or cosmide) Sequences of the vector can be mixed with your DNA sequence Before working with your DNA sequence, you should always clean it with VecScreen
 Sequences of the vector can be mixed with your DNA sequence Before working with your DNA sequence, you should always clean it with VecScreen")
15
VecScreen http://www.ncbi.nlm.nih.gov/VecScreen /VecScreen.html Runs a special version of Blast A system for quickly identifying segments of a nucleic acid sequence that may be of vector origin

20
What to do if hits found If hits are in the extremity, can just remove them If in the middle, or vectors are not what you are using, the safest thing is to throw the sequence away

21
Computing a Restriction Map It is possible to cut DNA sequences using restriction enzymes Each type of restriction enzyme recognizes and cuts a different sequence: EcoR1: GAATTC BamH1: GGATCC There are more than 900 different restriction enzymes, each with a different specificity The restriction map is the list of all potential cleavage sites in a DNA molecule You can compile a restriction map with www.firstmarket.com/cutter

22
Cannot get it work!

23
http://biotools.umassmed.edu/tacg4

25
Making PCR with a Computer Polymerase Chain Reaction (PCR) is a method for amplifying DNA PCR is used for many applications, including Gene cloning Forensic analysis Paternity tests PCR amplifies the DNA between two anchors These anchors are called the PCR primer
 is a method for amplifying DNA PCR is used for many applications, including Gene cloning Forensic analysis Paternity tests PCR amplifies the DNA between two anchors These anchors are called the PCR primer")
26
Designing PCR Primers PCR primes are typically 20 nucleotides long The primers must hybridize well with the DNA On biotools.umassmed.edu, find the best location for the primers: Most stable Longest extension

30
Analyzing DNA Composition DNA composition varies a lot Stability of a DNA sequence depends on its G+C content (total guanine and cytosine) High G+C makes very stable DNA molecules Online resources are available to measure the GC content of your DNA sequence Also for counting words and internal repeats
 High G+C makes very stable DNA molecules Online resources are available to measure the GC content of your DNA sequence Also for counting words and internal repeats")
31
http://helixweb.nih.gov/emboss/html/

32
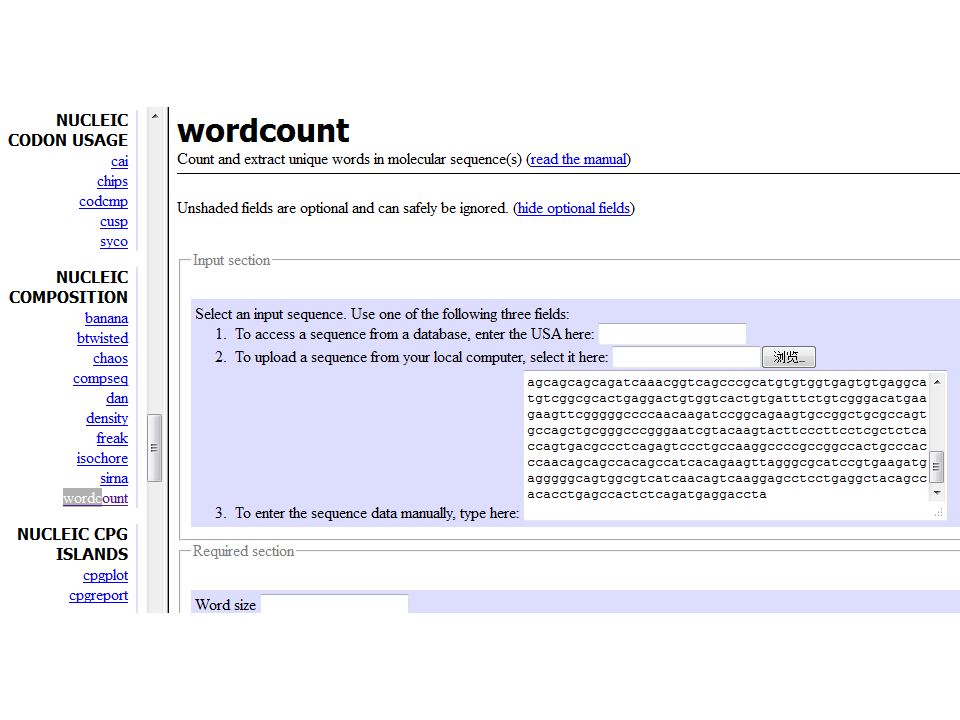
Counting words ATGGCTGACT A, T, G, G, C, T, G, A, C, T AT, TG, GG, GC, CT, TG, GA, AC, CT ATG, TGG, GGC, GCT, CTG, TGA, GAC, ACT

33
www.genomatix.de/cgi-bin/tools/tools.pl

34
EMBOSS servers European Molecular Biology Open Software Suite http://pro.genomics.purdue.edu/emboss/

38
ORF EMBOSS NCBI

41
ncbi.nlm.nih.gov/gorf/gorf.html

43
Internal repeats A word repeated in the sequence, long enough to not occur by chance Can be imperfect (regular expression) Dot plot is the best way to spot it
 Dot plot is the best way to spot it")
44
arbl.cvmbs.colostate.edu/molkit

46
Predicting Genes The most important analysis carried out on DNA sequences is gene prediction Gene prediction requires different methods for eukaryotes and prokaryotes Most gene-prediction methods use hidden Markov Models

47
Predicting Genes in Prokaryotic Genome In prokaryotes, protein-coding genes are uninterrupted No introns Predicting protein-coding genes in prokaryotes is considered a solved problem You can expect 99% accuracy

48
Finding Prokaryotic Genes with GeneMark GeneMark is the state of the art for microbial genomes GeneMark can Find short proteins Resolve overlapping genes Identify the best start codon Use exon.gatech.edu/GeneMark Click the “heutistic models”

49
Predicting Eukaryotic Genes Eukaryotic genes (human, for example) are very hard to predict Precise and accurate eukaryotic gene prediction is still an open problem ENSEMBL contains 21,662 genes for the human genome There may well be more genes than that in the genome, as yet unpredicted You can expect 70% accuracy on the human genome with automatic methods Experimental information is still needed to predict eukaryotic genes
 are very hard to predict Precise and accurate eukaryotic gene prediction is still an open problem ENSEMBL contains 21,662 genes for the human genome There may well be more genes than that in the genome, as yet unpredicted You can expect 70% accuracy on the human genome with automatic methods Experimental information is still needed to predict eukaryotic genes")
50
Finding Eukaryotic Genes with GenomeScan GenomeScan is the state of the art for eukaryotic genes GenomeScan works best with Long exons Genes with a low GC content It can incorporate experimental information Use genes.mit.edu/genomescan

51
Producing Genomic Data Until recently, sequencing an entire genome was very expensive and difficult Only major institutes could do it Today, scientists estimate that in 10 years, it will cost about $1000 to sequence a human genome With sequencing so cheap, assembling your own genomes is becoming an option How could you do it?

52
Sequencing and Assembling a Genome (I) To sequence a genome, the first task is to cut it into many small, overlapping pieces Then clone each piece
 To sequence a genome, the first task is to cut it into many small, overlapping pieces Then clone each piece")
53
Sequencing and Assembling a Genome (II) Each piece must be sequenced Sequencing machines cannot do an entire sequence at once They can only produce short sequences smaller than 1 Kb These pieces are called reads It is necessary to assemble the reads into contigs
 Each piece must be sequenced Sequencing machines cannot do an entire sequence at once They can only produce short sequences smaller than 1 Kb These pieces are called reads It is necessary to assemble the reads into contigs")
54
Sequencing and Assembling a Genome (III) The most popular program for assembling reads is PHRAP Available at www.phrap.org Other programs exist for joining smaller datasets For example, try CAP3 at pbil.univ-lyon1.fr/cap3.php
 The most popular program for assembling reads is PHRAP Available at Other programs exist for joining smaller datasets For example, try CAP3 at pbil.univ-lyon1.fr/cap3.php")
Similar presentations
















 BIOTECHNOLOGY.>")










E.g Biology Workbench ExPasy proteomics tools U. Mass. Med. School.Biotools.>")





