Download presentation
Presentation is loading. Please wait.

1
Chapter 3 Despair Inc.

2
POLYMERIZATION KINETICS REVIEW

3
[Reproduced from G. M. Kavanagh and S. B. Ross-Murphy, “Rheological characterisation of polymer gels”, Prog. Polym. Sci. 23, 533 (1998).] Characterization of polymers: Length and time scales are important
.] Characterization of polymers: Length and time scales are important.")
4
Polymer solubility In solvent, other polymer(s) or plasticizer Non-Newtonian properties: Viscosity, rheology Need to understand the configuration, conformation, and dynamics of macromolecules –Statistical mechanics Series of mathematical models Tied to experiment (viscosity, light scattering)
 or plasticizer Non-Newtonian properties: Viscosity, rheology Need to understand the configuration, conformation, and dynamics of macromolecules –Statistical mechanics Series of mathematical models Tied to experiment (viscosity, light scattering)")
5
Thermodynamics of Mixing: (Free energy) Entropy > 0, so –T S < 0 H can be negative or positive G = H -T S -T S For solution G < 0 Polymers are more sensitive to H
 Entropy > 0, so –T S < 0 H can be negative or positive G = H -T S -T S For solution G < 0 Polymers are more sensitive to H")
6
Spinoidal decomposition of a solution into two phases Quenching (rapid cooling) of solution Polymer rich & polymer poor phases Careful removal of polymer poor phase “microporous” Foams.
 of solution Polymer rich & polymer poor phases Careful removal of polymer poor phase microporous Foams.")
7
Any solution process is governed by the free energy relationship G = H - T S G < 0 polymer dissolves spontaneously S > 0 arising from increased conformational mobility of the polymer chain For a binary system, H mix : heat of mixing V 1,V 2 : molar volumes 1, 2 : volume fractions E 1, E 2 : energies of vaporization E 1 /V 1, E 2 /V 2 : cohesive energy densities

8
Enthalpy of Mixing H mix can be a positive or negative quantity –If A-A and B-B interactions are stronger than A-B interactions, then H mix > 0 (unmixed state is lower in energy) –If A-B interactions are stronger than pure component interactions, then H mix < 0 (solution state is lower in energy) An ideal solution is defined as one in which the interactions between all components are equivalent. As a result, H mix = H AB - (w A H A + w B H B ) = 0 for an ideal mixture In general, most polymer-solvent interactions produce H mix > 0, the exceptional cases being those in which significant hydrogen bonding between components is possible. –Predicting solubility in polymer systems often amounts to considering the magnitude of H mix > 0. –If the enthalpy of mixing is greater than T S mix, then we know that the lower Gibbs energy condition is the unmixed state.
 = 0 for an ideal mixture In general, most polymer-solvent interactions produce H mix > 0, the exceptional cases being those in which significant hydrogen bonding between components is possible. –Predicting solubility in polymer systems often amounts to considering the magnitude of H mix > 0. –If the enthalpy of mixing is greater than T S mix, then we know that the lower Gibbs energy condition is the unmixed state..")
9
1, 2 : solubility parameters H mix = V mix ( 1 - 2 ) 2 1 2 ∴ ( 1 - 2 ) 0; H mix small if ( 1 - 2 ) = 0 H mix = 0 then, G = - T S G < 0 Under this condition, solubility is governed solely by entropy effects i.e., For an ideal solvent s p Cohesive energy density ( E/V; 2 ) ● energy needed to remove a molecule from its nearest neighbors. ● analogous to heat of vaporization per volume for a volatile compound.

10
calculated directly from the latent heat of vaporization ( H vap ) For 1 (solvent) For 2 (polymer) estimated from group molar attraction constants G (due to their negligible vapor pressure) G G ● Consider intermolecular forces between molecular groups ● Derived from low M.W. compounds ● by Small heat of vaporization ● by Hoy vapor pressure measurements

11
Which set one uses is normally determined by the method used for determining 1 for the solvent. (V: molar volume, d: density, M: MW of repeat unit)
.")
12
Small’s system Hoy’s system For taking into account the strong intermolecular dipolar forces, solubility parameters d (dispersion forces) p (dipole-dipole attraction) H (hydrogen bonding) Hydrodynamic volume polymer size in solution ● Interaction between solvent and polymer molecules ● Chain branching ● Conformational effects (arising from the polarity and steric bulkiness of the substituent groups) Restricted rotation caused by resonance
 p (dipole-dipole attraction) H (hydrogen bonding) Hydrodynamic volume polymer size in solution ● Interaction between solvent and polymer molecules ● Chain branching ● Conformational effects (arising from the polarity and steric bulkiness of the substituent groups) Restricted rotation caused by resonance")
13
H mix and the Solubility Parameter The most popular predictor of polymer solubility is the solubility parameter, i. Originally developed to guide solvent selection in the paint and coatings industry, it is widely used in spite of its limitations. For regular solutions in which intermolecular attractions are minimal, H mix can be estimated through: where U 1,2 = internal energy change of mixing per unit volume, i = volume fraction of component i in the proposed mixture, i = solubility parameter of component i: (cal/cm 3 ) 1/2 Note that this formula always predicts H mix > 0, which holds only for regular solutions.
 1/2 Note that this formula always predicts H mix > 0, which holds only for regular solutions..")
14
The conditions of greatest polymer solubility exist when the solubility parameters of polymer and solvent match. If the polymer is crosslinked, it cannot dissolve but only swell as solvent penetrates the material. The solubility parameter of a polymer is therefore determined by exposing it to different solvents, and observing the at which swelling is maximized. Determining the Solubility Parameter

15
Solubility (Hildebrant) Parameters of Select Materials If < 1, polymer will dissolve
 Parameters of Select Materials If < 1, polymer will dissolve")
16
Solubility Parameters of Select Materials

17
Partial Miscibility of Polymers in Solvents Idealized representation of three generalized possibilities for the dependence of the Gibbs free energy of mixing, G m, of a binary mixture on composition (volume fraction of polymer, 2 ) at constant P and T. I. Total immiscibility; II. Partial miscibility; III. Total miscibility. Curve II represents the intermediate case of partial miscibility whereby the mixture will separate into two phases whose compositions ( ) are marked by the volume- fraction coordinates, 2 A and 2 B, corresponding to points of common tangent to the free-energy curve.
 are marked by the volume- fraction coordinates, 2 A and 2 B, corresponding to points of common tangent to the free-energy curve..")
18
Factors Influencing Polymer-Solvent Miscibility Based on the Flory-Huggins treatment of polymer solubility, we can explain the influence of the following variables on miscibility: 1. Temperature: The sign of G mix is determined by the Flory- Huggins interaction parameter, . As temperature rises, decreases thereby improving solubility. Upper solution critical temperature (UCST) behaviour is explained by Flory-Huggins theory, but LCST is not. 2. Molecular Weight: Increasing molecular weight reduces the configurational entropy of mixing, thereby reducing solubility. 3. Crystallinity:A semi-crystalline polymer has a more positive H mix = H AB - x A H A - x B H B due to the heat of fusion that is lost upon mixing.
 behaviour is explained by Flory-Huggins theory, but LCST is not. 2. Molecular Weight: Increasing molecular weight reduces the configurational entropy of mixing, thereby reducing solubility. 3. Crystallinity:A semi-crystalline polymer has a more positive H mix = H AB - x A H A - x B H B due to the heat of fusion that is lost upon mixing..")
19
Theta Temperature

22
Partial Miscibility of Polymers in Solvents Phase diagrams for the polystyrene-acetone system showing both UCSTs and LCSTs. Molecular weights of the polystyrene fractions are indicated.

23
Polymer-Solvent Miscibility 22 Phase diagrams for four samples of polystyrene mixed with cyclohexane plotted against the volume fraction of polystyrene. The molecular weight of each fraction is given. The dashed lines show the predictions of the Flory-Huggins theory for two of the fractions.

24
Practical Use of Polymer TDs Fractionation Consider solution in poor solvent of two polymers, p1 and p2. Flory-Huggins tells us that if p2 has higher molecular weight it should precipitate more readily than p1 add non-solvent until solution becomes turbid heat, cool slowly and separate precipitate finite drop in temperature always renders finite range of molecular weight insoluble some p2 will also remain soluble! T 2 volume fraction polymer p1 p2 2 phase cloudy 1 phase clear solution

25
Fractionation of Polymers using Precipitation

26
Industrial Relevance of Polymer Solubility

27
Polymer Solubility When two hydrocarbons such as dodecane and 2,4,6,8,10- pentamethyldodecane are combined, we (not surprisingly) generate a homogeneous solution: It is therefore interesting that polymeric analogues of these compounds, poly(ethylene) and poly(propylene) do not mix, but when combined produce a dispersion of one material in the other.
 generate a homogeneous solution: It is therefore interesting that polymeric analogues of these compounds, poly(ethylene) and poly(propylene) do not mix, but when combined produce a dispersion of one material in the other.")
28
Polymer solutions: Definitions Configuration: arrangement of atoms through bonds –Inter-change requires bond breaking and making (>200 kJ/mol) –Different compositions of polymers –Different isomers or stereochemistries Conformation: within the constraints of a configuration, the possible arrangement(s) of atoms in space –Interchange requires bond rotations, but no bonds are broken or made (can have non-bonding, ie. hydrogen bonding)
.")
29
What are the dimensions of a polymer? In a solution, in the solid state, in its melt, in vacuum? Fully stretched Polyethylene Polyethylene coil

30
Factors Affecting Macromolecule Dimensions

31
Polymer Conformations Flexible coil Rigid rod And everything in between

32
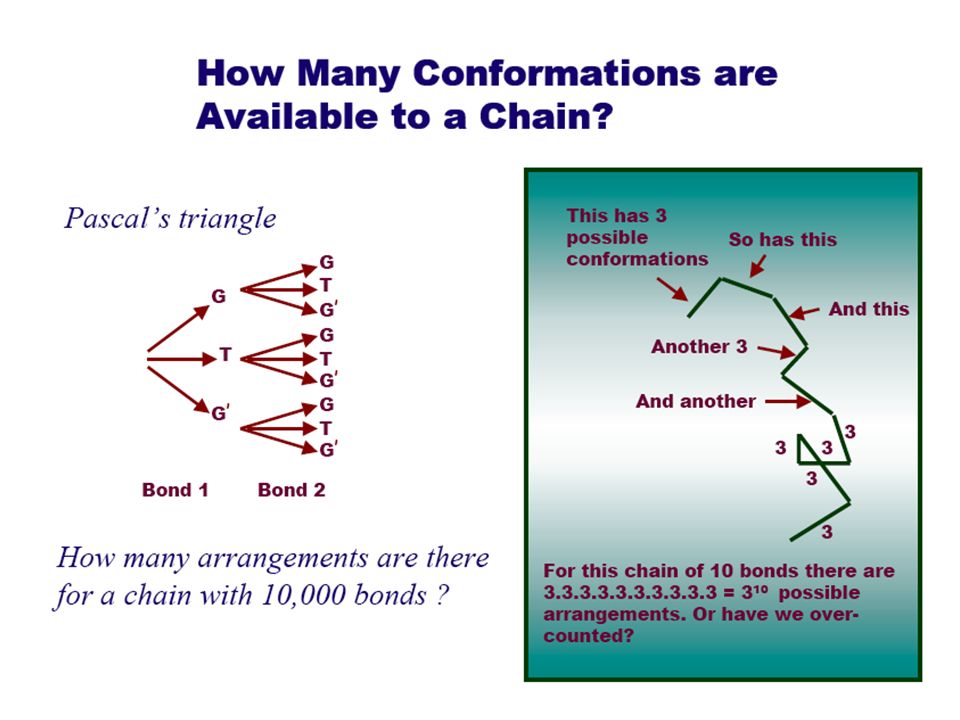
How to describe conformations with models: The freely jointed chain Simplest measure of a chain is the length along the backbone –For n monomers each of length l, the contour length is nl 123 n... l

33
First, Most Primative Model: Freely Jointed Chain Any bond angles & orientations are possible

34
For an isolated polymer in a solvent the end-to- end distance will change continuously due to molecular motion –But many conformation give rise to the same value of r, and some values of r are more likely than others e.g., Only one conformation with r = nl - a fully extended chain Many conformation have r = 0, (cyclic polymers) –Define the root mean square end-to-end distance –Permits statistical treatments A useful measure of the size of macromolecules: end-to-end distance r Free rotation model: Infinite number of conformations But we still don’t know the shape; need something more
 –Define the root mean square end-to-end distance –Permits statistical treatments A useful measure of the size of macromolecules: end-to-end distance r Free rotation model: Infinite number of conformations But we still don’t know the shape; need something more")
36
Example of a “low resolution” polymer coil RW. Contour length = l ct, unit vectors to describe the length = e(l), = vectors that connect the junction points on the chain, r = end-to-end distance of the chain A freely jointed chain in 2D from a random walk of 50 steps. Random Walk Model for linear polymers # of steps based on number atoms in chain Complications: excluded volume & steric limitations
, = vectors that connect the junction points on the chain, r = end-to-end distance of the chain A freely jointed chain in 2D from a random walk of 50 steps. Random Walk Model for linear polymers # of steps based on number atoms in chain Complications: excluded volume & steric limitations.")
37
An ideal polymer chain with 10 6 repeat units (not unusual), each unit about 6Å will have: a rms end-to-end distance R of 600 nm a contour length of 600 μm What does this all mean??
, each unit about 6Å will have: a rms end-to-end distance R of 600 nm a contour length of 600 μm What does this all mean")
38
A better measure of Polymer dimensions: Radius of Gyration of a Polymer Coil The radius of gyration R g is defined as the RMS distance of the collection of atoms from their common centre of gravity. For a solid sphere of radius R; For a polymer coil with rms end-to-end distance R ; R Can be related directly to statistical distributions from polymer characterizations RgRg

39
Restrict the bond angles to what we see in the polymer: Valence Angle Model

40
Valence angle model Simplest modification to the freely jointed chain model –Introduce bond angle restrictions –Allow free rotation about bonds –Neglecting steric effects (for now) If all bond angles are equal to , indicates that the result is for the valence angle model E.g. for polyethylene = 109.5° and cos ~ -1/3, hence,

41
Finite Number of Conformations due to torsional & steric interactions: Restricted Rotation Angle The energy barrier between gauche and trans is about 2.5 kJ/mol RT~8.31*300 J/mol~2.5 kJ/mol

47
Equivalent Freely Jointed Chain Model C ∞ is a function of the stiffness of the chain. Higher is stiffer C∞C∞

48
In general –where is the steric parameter, which is usually determined for each polymer experimentally –A measure of the stiffness of a chain is given by the characteristic ratio –C typically ranges from 5 - 12 Steric parameter and the characteristic ratio

49
Polymer Conformations Flexible coil Rigid rod 1/2 ~ N 1/2 1/2 ~ N The shape of the polymers can therefore only be usefully described statistically.

50
Calculate size of macromolecules!

51
Freely jointed chain, valence angle and rotational isomeric states models all ignore –long range intramolecular interactions (e.g. ionic polymers) –polymer-solvent interactions Such interactions will affect –Define where is the expansion parameter Excluded volume
 –polymer-solvent interactions Such interactions will affect –Define where is the expansion parameter Excluded volume.")
52
Space filling? The random walk and the steric limitations makes the polymer coils in a polymer melt or in a polymer glass “expanded”. However, the overlap between molecules ensure space filling

53
Expansion Factors: Measure of solvent -Polymer Interactions

54
: mean-square average distance between chain ends for a linear polymer : square average radius of gyration about the center of gravity for a branched polymer Better solvent stronger interaction between solvent and polymer larger hydrodynamic volume r 0, s 0 : unperturbed dimension (i.e. the size of the macromolecule exclusive of solvent effects) : an expansion factor
 : an expansion factor.")
55
(for a linear polymer) greater better solvent = 1 ideal statistical coil Solubility vary with temperature in a given solvent is temperature dependent Theta (θ) temperature (Flory temperature) ● For a given polymer in a given solvent, the lowest temperature at which = 1 ● state / solvent Polymer in a state having a minimal solvation effect on the brink of becoming insoluble further diminution of solvation effect polymer precipitation
 greater better solvent = 1 ideal statistical coil Solubility vary with temperature in a given solvent is temperature dependent Theta (θ) temperature (Flory temperature) ● For a given polymer in a given solvent, the lowest temperature at which = 1 ● state / solvent Polymer in a state having a minimal solvation effect on the brink of becoming insoluble further diminution of solvation effect polymer precipitation")
56
The expansion parameter r Solvent Effects on the Macromolecules r depends on balance between i) polymer-solvent and ii) polymer-polymer interactions –If polymer-polymer are more favourable than polymer-solvent r < 1 Chains contract Solvent is poor –If polymer-polymer are less favourable than polymer-solvent r > 1 Chains expand Solvent is good –If these interactions are equivalent, we have theta condition r = 1 Same as in amorphous melt
 polymer-solvent and ii) polymer-polymer interactions –If polymer-polymer are more favourable than polymer-solvent r < 1 Chains contract Solvent is poor –If polymer-polymer are less favourable than polymer-solvent r > 1 Chains expand Solvent is good –If these interactions are equivalent, we have theta condition r = 1 Same as in amorphous melt")
57
The theta temperature For most polymer solutions r depends on temperature, and increases with increasing temperature At temperatures above some theta temperature, the solvent is good, whereas below the solvent is poor, i.e., What determines whether or not a polymer is soluble? T > r > 1 T = r = 1 T < r < 1 Often polymers will precipitate out of solution, rather than contracting

58
Solubility of Polymers Encyclopedia of Polymer Science, Vol 15, pg 401 says it best... A polymer is often soluble in a low molecular weight liquid if: the two components are similar chemically or are so constituted that specific attractive interactions such as hydrogen bonding take place between them; the molecular weight of the polymer is low; the bulk polymer is not crystalline; the temperature is elevated (except in systems with LCST). The method of solubility parameters can be useful for identifying potential solvents for a polymer. Some polymers that are not soluble in pure liquids can be dissolved in a multi- component solvent mixture. Binary polymer-polymer mixtures are usually immiscible except when they possess a complementary dissimilarity that leads to negative heats of mixing.
. The method of solubility parameters can be useful for identifying potential solvents for a polymer. Some polymers that are not soluble in pure liquids can be dissolved in a multi- component solvent mixture. Binary polymer-polymer mixtures are usually immiscible except when they possess a complementary dissimilarity that leads to negative heats of mixing..")
59
Another way of looking at Solubility: Thermodynamics of Mixing G mix < 0 G mix > 0 immiscible blend A-B solution G mix (Joules/gram) is defined by: G mix = H mix -T S mix where H mix = H AB - (x A H A + x B H B ) S mix = S AB - (x A S A + x B S B ) and x A, x B are the mole fractions of each material. m A moles m B moles material A material B + Polymer-solvent: volume fraction of polymers

60
Ethanol(1) / chloroform(2) at 50ºC Ethanol(1) / n-heptane(2) at 50ºC Ethanol(1) / water(2) at 50ºC Thermodynamics of Mixing: Small Molecules
 / chloroform(2) at 50ºC Ethanol(1) / n-heptane(2) at 50ºC Ethanol(1) / water(2) at 50ºC Thermodynamics of Mixing: Small Molecules")
61
Solvent-Solvent Solutions vs Polymer-Solvent Solutions Solvents can easily replace one another. Polymers are thousands of solvent sized monomers connected together. Monomers can not be placed randomly! Entropy of mixing is generally positive Enthalpy is also often positive: smaller the better Ideal = 0

62
Flory-Huggins Theory Flory-Huggins theory, originally derived for small molecule systems, was expanded to model polymer systems by assuming the polymer consisted of a series of connected segments, each of which occupied one lattice site. Assuming segments are randomly distributed and that all lattice sites are occupied, the free energy of mixing per mole of lattice sites is G mix =RT[( A /N A )ln A +( B /N B )ln B + FH A B ] (3) i is the volume fraction of polymer i N i is the number of segments in polymer i HF is the Flory-Huggins interaction parameter
ln A +( B /N B )ln B + FH A B ] (3) i is the volume fraction of polymer i N i is the number of segments in polymer i HF is the Flory-Huggins interaction parameter.")
63
Gibbs Energy of Mixing: Flory-Huggins Theory Combining expressions for the enthalpy and entropy of mixing generates the free energy of mixing: The two contributions to the Gibbs energy are configurational entropy as well as an interaction entropy and enthalpy (characterized by ; “chi” pronounced “Kigh”). Note that for complete miscibility over all concentrations, for the solute-solvent pair at the T of interest must be less than 0.5. If > 0.5, then G mix > 0 and phase separation occurs If < 0.5, then G mix < 0 over the whole composition range. The temperature at which = 0.5 is the theta temperature.

64
For a mixture of polymer and solvent or two polymers Mixing is always entropically good.

65
Gibbs Energy of Mixing: Flory-Huggins Theory Combining expressions for the enthalpy and entropy of mixing generates the free energy of mixing: The two contributions to the Gibbs energy are configurational entropy as well as an interaction entropy and enthalpy (characterized by ; “chi” pronounced “Kigh”). Note that for complete miscibility over all concentrations, for the solute-solvent pair at the T of interest must be less than 0.5. If > 0.5, then G mix > 0 and phase separation occurs If < 0.5, then G mix < 0 over the whole composition range. The temperature at which = 0.5 is the theta temperature.

66
Case 2: Exothermic mixing (negative H) (2E AB < E AA + E BB ) < 0.5 No Phase separation: Completely Miscible
 (2E AB < E AA + E BB ) < 0.5 No Phase separation: Completely Miscible")
67
Case 3: Endothermic mixing (positive H) (2E AB > E AA + E BB ) > 0.5 Partial Miscibility: two phases of mixed compositions
 (2E AB > E AA + E BB ) > 0.5 Partial Miscibility: two phases of mixed compositions")
68
Start with homogeneous mixture composition c at free energy of F. Splits into two phases.

69
Blends are not easy to make: Blends are not easy to discover-often copolymers are needed to make blend with another homopolymer Phase separation gets out of hand in immiscible systems without compatibilizers. Some blends must be made with solvent.

70
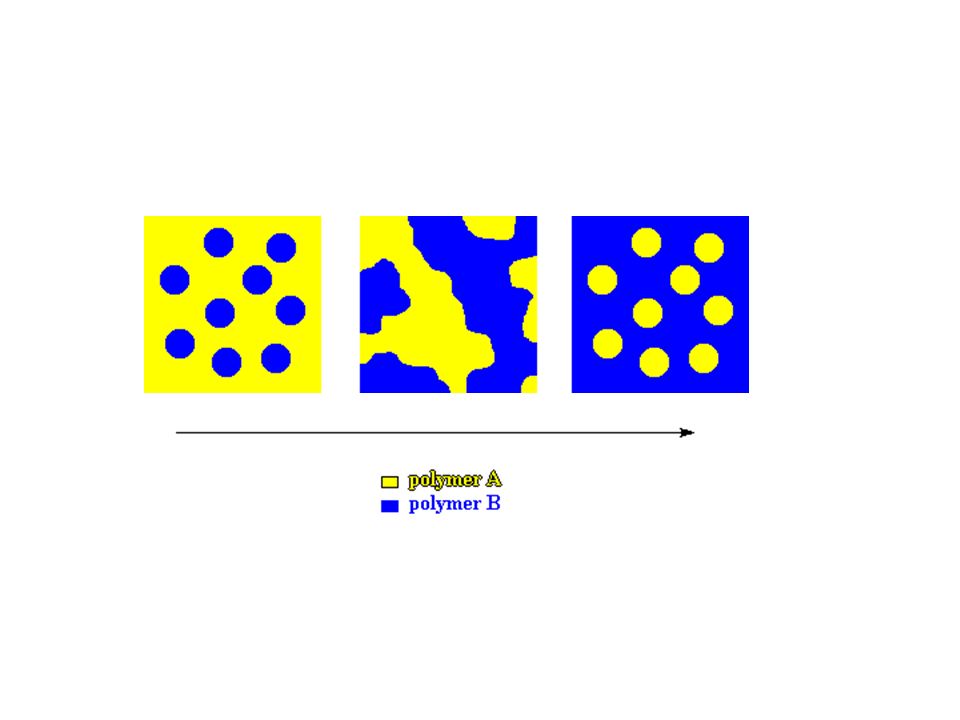
Polymer Mixture Immiscible

71
Some miscible blends

72
Polymer blend poly(ethylene terephthalate) with poly(butylene terephthalate)
 with poly(butylene terephthalate)")
73
Some miscible blends PMMA & poly(vinylidene fluoride)
")
74
Some miscible blends Mini Cooper / Cooper S, radiator grille & PETE

75
with

76
Immiscible Blends

80
1:6 (PTV:PCBM) 1:4 (PTV:PCBM) 1:3 (PTV:PCBM) 1:2 (PTV:PCBM) 1:10 (PTV:PCBM)
 1:4 (PTV:PCBM) 1:3 (PTV:PCBM) 1:2 (PTV:PCBM) 1:10 (PTV:PCBM)")
81
Random: AABABBAAABABABB Alternating: ABABABABABABABA Block: AAAAAAAABBBBBBB 1.Linear polymer: copolymers Morphology of diblock Copolymers:

82
PMMA-PS-PB ternary blend

84
Dilute Solution Viscosity The “strength” of a solvent for a given polymer not only effects solubility, but the conformation of chains in solution. –A polymer dissolved in a “poor” solvent tends to aggregate while a “good” solvent interacts with the polymer chain to create an expanded conformation. –Increasing temperature has a similar effect to solvent strength. The viscosity of a polymer solution is therefore dependent on solvent strength. Consider Einstein’s equation: s (1+2.5 ) where is the viscosity s is the solvent viscosity and is the volume fraction of dispersed spheres.
 where is the viscosity s is the solvent viscosity and is the volume fraction of dispersed spheres..")
86
Molecular Weight and Polymer Solutions Number Average and Weight Average Molecular Weight Determine M. W. of small molecules Mass spectrometry Cryoscopy (freezing-point depression) Ebulliometry (boiling-point elevation) Titration Determine M. W. of polymers Osmometry M n Light scattering M w Ultracentrifugation M w End-group analysis M n
 Ebulliometry (boiling-point elevation) Titration Determine M. W. of polymers Osmometry M n Light scattering M w Ultracentrifugation M w End-group analysis M n.")
87
Measurement of Number Average Molecular Weight End-group analysis Titration polyester (-COOH, -OH), polyamide (-C(O)NH-), polyurethanes( isocyanate), epoxy polymer (epoxide), acetyl-terminated polyamide (acetyl) Elemental analysis Radioactive labeling UV / NMR / (IR) Upper limit M. W. 50,000 (due to the low concentraction of end groups) preferred range: 5,000-10,000 Not applicable to branched polymers Analysis is meaningful only when the mechanisms of initiation and termination are well understood
 preferred range: 5,000-10,000 Not applicable to branched polymers Analysis is meaningful only when the mechanisms of initiation and termination are well understood.")
88
Membrane Osmometry ● based on colligative properties ● most useful method for M n (range: 50,000 to 2,000,000) ● major error source arising from the loss of low MW species ● obtained M n values are generally higher than other colligative measurement method Static equilibrium method measure the hydrostatic head ( h) after equilibrium Dynamic equilibrium method measure the counter pressure needed to maintain equal liquid levels h
 ● major error source arising from the loss of low MW species ● obtained M n values are generally higher than other colligative measurement method Static equilibrium method measure the hydrostatic head ( h) after equilibrium Dynamic equilibrium method measure the counter pressure needed to maintain equal liquid levels h")
89
Osmotic pressure ( ) related to by the van’t Hoff equation extrapolated to zero concentration (C; g/L) to obtain the intercept A 2 = v 2 V -1 (0.5 – )
 related to by the van’t Hoff equation extrapolated to zero concentration (C; g/L) to obtain the intercept A 2 = v 2 V -1 (0.5 – )")
90
Cryoscopy and Ebulliometry Thermodynamic relationships for freezing-point depression for boiling-point elevation Limited by the sensitivity of measuring T f, T b As MW higher; T f, T b smaller Upper limit - Mn = 40,000 Preferred - M n < 20,000

91
Vapor Pressure Osmometry ● For M n < 25,000 ● No membrane needed Thermodynamic principle similar to membrane osmometry Measurement method → Add a drop of solvent and solution, on a pair of matched themistors, in an insulated chamber saturated with solvent vapor then, → Condensation heats the solution thermistor (until the vapor pressure of the solution equal to that of the pure solvent) Temperature change is measured (by resistance change of the thermistor) and related to solution molality : heat of vaporization per gram (solvent) m : molarity
 Temperature change is measured (by resistance change of the thermistor) and related to solution molality : heat of vaporization per gram (solvent) m : molarity")
92
Mass Spectrometry MALDI-MS (MALDI-TOF) MALDI-MS (matrix-assisted laser desorption ionization mass spectrometry) (TOF - time-of-flight) ● Imbeds polymer in a matrix of low MW organic compound ● Irradiates the matrix with UV laser ● Matrix transfer the absorbed energy to polymer and vaporize the polymer ● Integrated peak areas the number of ions ● M n, M w can be calculated Soft ionization method field desorption (FD-MS) laser desorption (LD-MS) electrospray ionization (ESI-MS)
 MALDI-MS (matrix-assisted laser desorption ionization mass spectrometry) (TOF - time-of-flight) ● Imbeds polymer in a matrix of low MW organic compound ● Irradiates the matrix with UV laser ● Matrix transfer the absorbed energy to polymer and vaporize the polymer ● Integrated peak areas the number of ions ● M n, M w can be calculated Soft ionization method field desorption (FD-MS) laser desorption (LD-MS) electrospray ionization (ESI-MS)")
93
Refractive Index Measurement Most suitable for low MW polymers n Measurement of Weight Average Molecular Weight (Mw) Light Scattering Most widely used method for measuring absolute Light loses energy by absorption, conversion to heat, and scattering Light scattering is caused by inhomogeneous distribution of molecules due to the irregular change in density and refractive index (because of the fluctuation in composition) Scattering intensity depends on concentration, size and polarizability of the molecules Refractive index also depends on concentration and amplitude of vibration
 Light Scattering Most widely used method for measuring absolute Light loses energy by absorption, conversion to heat, and scattering Light scattering is caused by inhomogeneous distribution of molecules due to the irregular change in density and refractive index (because of the fluctuation in composition) Scattering intensity depends on concentration, size and polarizability of the molecules Refractive index also depends on concentration and amplitude of vibration")
94
LS adds optical effects Size q = 0 in phase I s maximum q > 0 out of phase, I s goes down

95
Scattering causes turbidity ( ) n 0 : solvent refractive index : wavelength of light N 0 : Avogadro’s number n: refractive index of solution dn/dc (specific refractive increment) is a constant for a given polymer, solvent, and temperature measured from the slope of n vs. c Debye equation (A 2 : the second virial coefficient) =
 =.")
96
As molecular size approaches the wavelength of the light Interference between scattered light coming from different parts occurs Correction is needed Turbidity ( or R ) associated with large particles measured at different concentrations (c) and angles ( ) P( ): Particle scattering factor P( ) is a function of ; depending on the shape of molecule in solution For a monodisperse of randomly coiling polymers
 associated with large particles measured at different concentrations (c) and angles ( ) P( ): Particle scattering factor P( ) is a function of ; depending on the shape of molecule in solution For a monodisperse of randomly coiling polymers")
97
Zimm Plot Plot vs. k is an arbitrary constant Extrapolated to both zero concentration and zero angle intercept = (P( ) = 1 at c = 0 and = 0)
 = 1 at c = 0 and = 0).")
98
Light scattering photometer Suitable range of M w : 10,000 - 10,000,000 Dust-free solution is required Light source Old: high-pressure Hg lamps plus filter New: laser

100
Most expensive Extensively used with proteins Determining Ultracentrifugation Sedimentation rate is proportional to molecular mass Distributed according to size along the perpendicular direction Concentration gradients within the polymer solution are observed by refractive index measurements and interferometry

101
Simplest and most widely used Not an absolute number Can be calibrated with the absolute MW (by light scattering) of fractionated polymer samples Typical conditions: 0.5 g/100 mL; 30.0±0.01 ℃ Viscometry Viscometers: (a) Ubbelohde (b) Cannon-Fenske Ubbelohde type more convenient not necessary to have exact volumes of solution additional solvent can be added (for dilution) dust particles can greatly alter the flow time (via the capillary tube)
 of fractionated polymer samples Typical conditions: 0.5 g/100 mL; 30.0±0.01 ℃ Viscometry Viscometers: (a) Ubbelohde (b) Cannon-Fenske Ubbelohde type more convenient not necessary to have exact volumes of solution additional solvent can be added (for dilution) dust particles can greatly alter the flow time (via the capillary tube)")
102
Relative viscosity (viscosity ratio), rel ● ratio of solution viscosity to solvent viscosity ● viscosity units (poises) or flow times cancel out (→ dimensionless) Viscosity Specific viscosity ( sp ) ● fractional increase in viscosity ● dimensionless Inherent viscosity ( inh ; dL/g) ● determined only by a single solution at a specific concentration ● an approximate indication of MW Intrinsic viscosity ([ ]; dL/g) ● most useful ● eliminating the concentration effect ● by dividing sp with C and then extrapolating to zero C
![Relative viscosity (viscosity ratio), rel ● ratio of solution viscosity to solvent viscosity ● viscosity units (poises) or flow times cancel out (→ dimensionless) Viscosity Specific viscosity ( sp ) ● fractional increase in viscosity ● dimensionless Inherent viscosity ( inh ; dL/g) ● determined only by a single solution at a specific concentration ● an approximate indication of MW Intrinsic viscosity ([ ]; dL/g) ● most useful ● eliminating the concentration effect ● by dividing sp with C and then extrapolating to zero C](http://images.slideplayer.com/23/6864671/slides/slide_102.jpg "Relative viscosity (viscosity ratio), rel ● ratio of solution viscosity to solvent viscosity ● viscosity units (poises) or flow times cancel out (→ dimensionless) Viscosity Specific viscosity ( sp ) ● fractional increase in viscosity ● dimensionless Inherent viscosity ( inh ; dL/g) ● determined only by a single solution at a specific concentration ● an approximate indication of MW Intrinsic viscosity ([ ]; dL/g) ● most useful ● eliminating the concentration effect ● by dividing sp with C and then extrapolating to zero C")
103
Common NameIUPAC Name Relative viscosityViscosity ratio Specific viscosity - Reduced viscosityViscosity number Inherent viscosity Logarithmic viscosity number Intrinsic viscosity Limiting viscosity number

104
viscosity average molecular weight (Mark-Houwink-Sakurada) plot log [ ] vs log (or log ) measure a and K in general, << (closer to) better results are obtained if the MW of fractionated samples are used (here, ) ● typically, a 0.5 - 0.8, K 10 -3 - 0.5 ● for randomly coiled polymer in a solvent a = 0.5; < for a rodlike extended chain polymer a = 1.0;
![viscosity average molecular weight (Mark-Houwink-Sakurada) plot log [ ] vs log (or log ) measure a and K in general, << (closer to) better results are obtained if the MW of fractionated samples are used (here, ) ● typically, a 0.5 - 0.8, K - 0.5 ● for randomly coiled polymer in a solvent a = 0.5; < for a rodlike extended chain polymer a = 1.0;](http://images.slideplayer.com/23/6864671/slides/slide_104.jpg "viscosity average molecular weight (Mark-Houwink-Sakurada) plot log [ ] vs log (or log ) measure a and K in general, << (closer to) better results are obtained if the MW of fractionated samples are used (here, ) ● typically, a 0.5 - 0.8, K - 0.5 ● for randomly coiled polymer in a solvent a = 0.5; < for a rodlike extended chain polymer a = 1.0;")
105
In polymers a values have solvent dependence

106
[η] = KM a Linear polymers: = 0.5-1.0 Rigid rod: = 1.0-2.0 Branched polymers: = 0.21-0.28 Rigid spherical particles: = 0
![[η] = KM a Linear polymers: = Rigid rod: = Branched polymers: = Rigid spherical particles: = 0](http://images.slideplayer.com/23/6864671/slides/slide_106.jpg "[η] = KM a Linear polymers: = Rigid rod: = Branched polymers: = Rigid spherical particles: = 0")
107
Complication in applying Mark-Houwink-Sakurada relationship chain branching broad MWD sample solvation of polymer backbone sequences (alternating or block) chain entanglement (when MW is extremely large) Viscosity measurements based on mechanical shearing for concentrated polymer solutions or undiluted polymer system more applicable to the flow properties of polymers
 chain entanglement (when MW is extremely large) Viscosity measurements based on mechanical shearing for concentrated polymer solutions or undiluted polymer system more applicable to the flow properties of polymers")
108
Dynamic Viscosity Viscosity = Pa s = kg m s -1 Force/Area = Pa m -1 = kg m -1 s -2 Force: Pa = N m -2 = kg m s -2 1 Pa s = 10 Poise 1 centipoise = 1 millipascal second (mPa s) Other units (Poise = dyne s cm -2 ) Informally: resistance to flow Formally: Ratio of Shearing stress (F/A) to the velocity gradient (dv x /d z )
 Other units (Poise = dyne s cm -2 ) Informally: resistance to flow Formally: Ratio of Shearing stress (F/A) to the velocity gradient (dv x /d z )")
109
EXAMPLES Water 1 mPa s Honey 10,000 mPa s Peanut Butter 200,000 mPa s

110
Kinematic viscosity is the dynamic viscosity divided by the density (typical units cm 2 /s, Stokes, St).
.")
112
Flow Regimes of Typical Polymer Processing High-speed coating Injection molding Lubrication Sedimentation Rolling Pipe flow Extrusion Spraying Chewing Typical viscosity curve of a polyproprene (PP), melt flow rate (230 C/2.16 Kg) of 8 g/10 min at 230 C with indication of the shear rate regions of different conversion techniques. [Reproduced from M. Gahleitner, “Melt rheology of polyolefins”, Prog. Polym. Sci. 26, 895 (2001).]
.].")
Similar presentations




























. What happens to a.>")



 a. two atoms of hydrogen b. One atom of oxygen c. Bent structure.>")