Download presentation
Presentation is loading. Please wait.

1
Cancer Clinical Trials in the Genomic Era Richard Simon, D.Sc. Chief, Biometric Research Branch National Cancer Institute http://brb.nci.nih.gov

2
Prognostic biomarkers Measured before treatment to indicate long-term outcome for patients untreated or receiving standard treatment May reflect both disease aggressiveness and effect of standard treatment Used to determine who needs more intensive treatment Predictive biomarkers Measured before treatment to identify who will benefit from a particular treatment

3
Endpoint Measured before, during and after treatment to monitor pace of disease and treatment effect Pharmacodynamic (phase 0-1) Does drug hit target Intermediate response (phase 2) Does drug have anti-tumor effect Surrogate for clinical outcome (phase 3)
 Does drug hit target Intermediate response (phase 2) Does drug have anti-tumor effect Surrogate for clinical outcome (phase 3)")
4
Prognostic & Predictive Biomarkers Single gene or protein measurement Scalar index or classifier that summarizes contributions of multiple genes

5
Prognostic & Predictive Biomarkers in Genomic Oncology Many cancer treatments benefit only a minority of patients to whom they are administered Being able to predict which patients are likely to benefit can Help patients get an effective treatment Help control medical costs Improve the success rate of clinical drug development

6
Validation = Fitness for Intended Use

7
Biomarker Validity Analytical validity Measures what it’s supposed to Reproducible and robust Clinical validity (correlation) It correlates with something clinically Medical utility Actionable resulting in patient benefit
 It correlates with something clinically Medical utility Actionable resulting in patient benefit")
8
Clinical Utility Biomarker informs action that benefits patient by improving treatment decisions Identify patients who have very good prognosis on standard treatment and do not require more intensive regimens Identify patients who are likely or unlikely to benefit from a specific regimen

9
Objective: Use biomarkers to Develop effective treatments Know who needs these treatments and who benefits from them 9

10
Prognostic markers There is an enormous published literature on prognostic markers in cancer. Very few prognostic markers (factors) are recommended for measurement by ASCO, are approved by FDA or are reimbursed for by payers. Very few play a role in treatment decisions.
 are recommended for measurement by ASCO, are approved by FDA or are reimbursed for by payers. Very few play a role in treatment decisions..")
11
Prognostic Biomarkers Can be Therapeutically Relevant <10% of node negative ER+ breast cancer patients require or benefit from the cytotoxic chemotherapy that they receive

12
OncotypeDx Recurrence Score Intended use: Patients with node negative estrogen receptor positive breast cancer who are going to receive an anti-estrogen drug following local surgery/radiotherapy Identify patients who have such good prognosis that they are unlikely to derive much benefit from adjuvant chemotherapy

13
Selected patients relevant for the intended use Analyzed the data to see if the recurrence score identified a subset with such good prognosis that the absolute benefit of chemotherapy would at best be very small in absolute terms Used an analytically validated test

15
Major problems with prognostic studies of gene expression signatures Inadequate focus on intended use Reporting highly biased estimates of predictive value

16
Major problems with prognostic studies of gene expression signatures Inadequate focus on intended use Cases selected based on availability of specimens rather than for relevance to intended use Heterogeneous sample of patients with mixed stages and treatments. Attempt to disentangle effects using regression modeling Too a great a focus on which marker is prognostic or independently prognostic, not whether the marker is effective for intended use

17
Goodness of fit is not a proper measure of predictive accuracy Odds ratios and hazards ratios are not proper measures of prediction accuracy Statistical significance of regression coefficients are not proper measures of predictive value

18
Goodness of Fit vs Prediction Accuracy For p>n problems, fit of a model to the same data used to develop it is no evidence of prediction accuracy for independent data

20
Validation of Prognostic Model Completely independent validation dataset Splitting dataset into training and testing sets Evaluate 1 completely specified model on test set Cross-validation

21
Leave-one-out Cross Validation for Classifier of Two Classes Full dataset P={1,2,…,n} Omit case 1 V 1 ={1}; T 1 ={2,3,…,n} Develop classifier using training set T 1 Classify cases in V 1 and count whether classification is correct or not Repeat for case 2,3,… Total number of mis-classified cases

22
Complete cross Validation Cross-validation simulates the process of separately developing a model on one set of data and predicting for a test set of data not used in developing the model All aspects of the model development process must be repeated for each loop of the cross-validation Feature selection Tuning parameter optimization

23
Cross Validation The cross-validated estimate of misclassification error is an estimate of the prediction error for the model fit applying the specified algorithm to full dataset

24
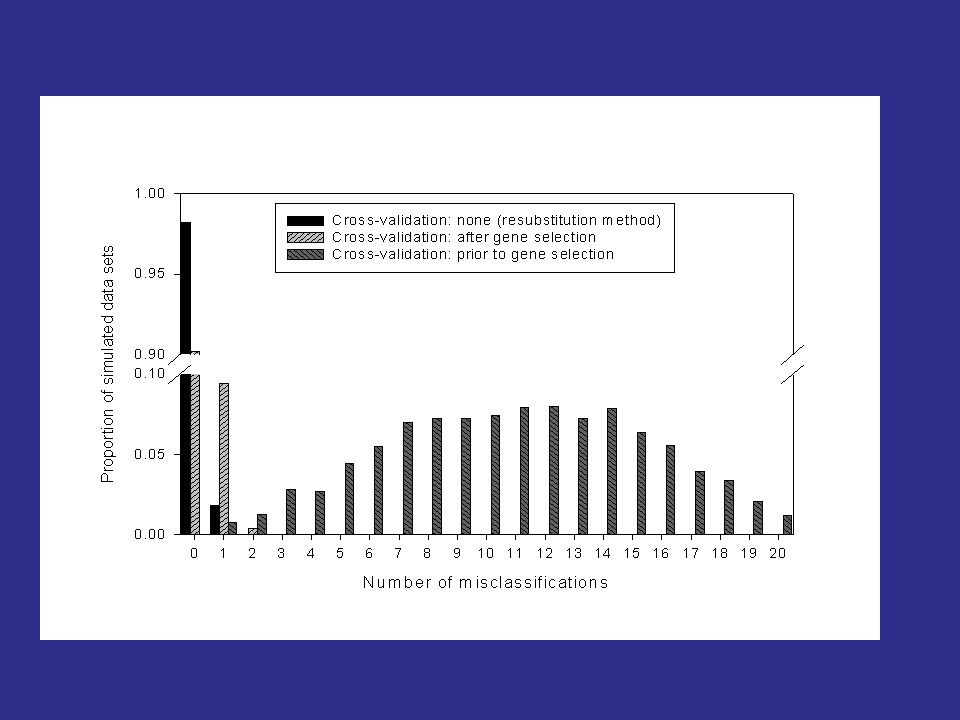
Prediction on Simulated Null Data Simon et al. J Nat Cancer Inst 95:14, 2003 Generation of Gene Expression Profiles 20 specimens (P i is the expression profile for specimen i) Log-ratio measurements on 6000 genes P i ~ MVN(0, I 6000 ) Can we distinguish between the first 10 specimens (Class 1) and the last 10 (Class 2)? Prediction Method Compound covariate predictor built from the log-ratios of the 10 most differentially expressed genes.
 Log-ratio measurements on 6000 genes P i ~ MVN(0, I 6000 ) Can we distinguish between the first 10 specimens (Class 1) and the last 10 (Class 2). Prediction Method Compound covariate predictor built from the log-ratios of the 10 most differentially expressed genes..")
26
Cross-validation Estimate of Prediction Error 26

28
Partition data set D into K equal parts D 1,D 2,...,D K First training set T 1 =D-D 1 Develop completely specified prognostic model M 1 using only data T 1 eg Using M 1, compute prognostic score for cases in D 1 Develop model M 2 using only T 2 and then score cases in D 2

29
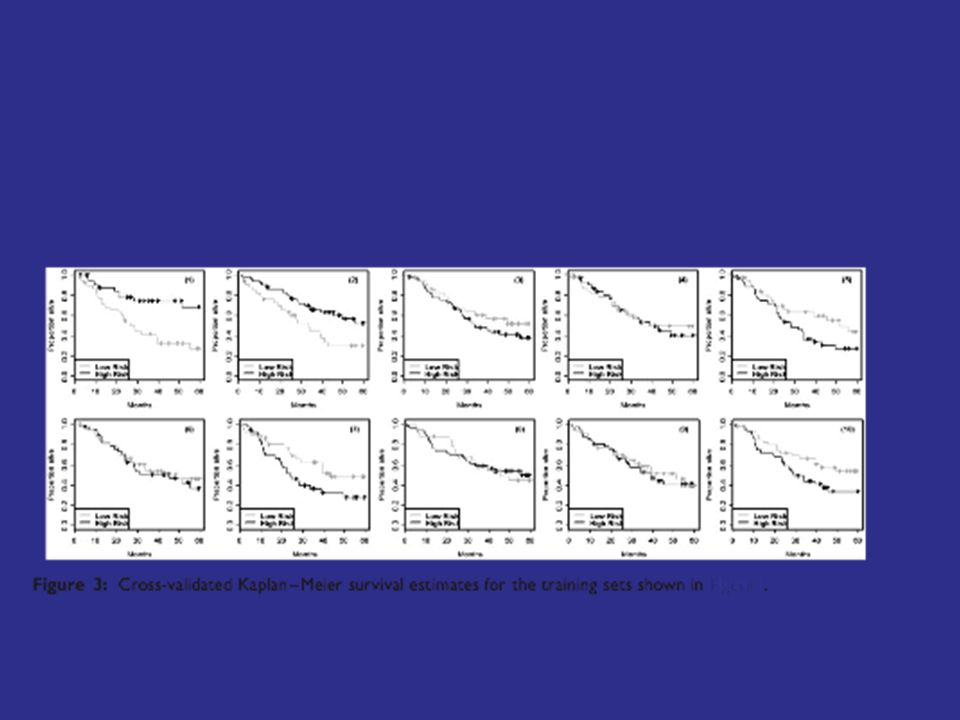
Repeat for... T K -> M K -> D K Group patients into 2 or more risk groups based on their cross-validated scores Calculate Kaplan-Meier survival curve for each risk-group

31
To evaluate significance, the log-rank test cannot be used for cross-validated Kaplan-Meier curves because the survival times are not independent

32
Statistical significance can be properly evaluated by approximating the null distribution of the cross-validated log-rank statistic Permute the survival times and repeat the entire cross-validation procedure to generate new cross-validated K-M curves for low risk and high risk groups – Compute log-rank statistic for the curves Repeat for many sets of permutations

33
Predictive Biomarkers Cancers of a primary site often represent a heterogeneous group of diverse molecular entities which vary fundamentally with regard to the oncogenic mutations that cause them their responsiveness to specific drugs

34
In most positive phase III clinical trials comparing a new treatment to control, most of the patients treated with the new treatment did not benefit. Adjuvant breast cancer: 70% long-term disease-free survival on control. 80% disease-free survival on new treatment. 70% of patients don’t need the new treatment. Of the remaining 30%, only 1/3 rd benefit.

35
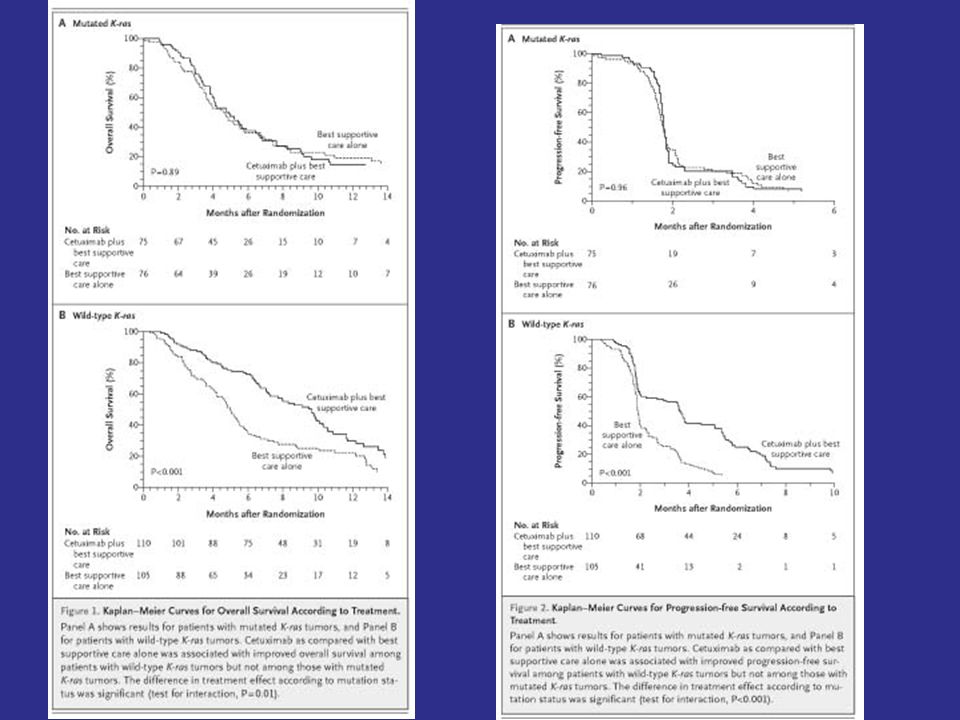
Predictive Biomarkers Estrogen receptor over-expression in breast cancer Anti-estrogens, aromatase inhibitors HER2 amplification in breast cancer Trastuzumab, Lapatinib OncotypeDx gene expression recurrence score in N+ ER+ breast cancer Low score -> not responsive to chemotherapy KRAS in colorectal cancer WT KRAS = cetuximab or panitumumab EGFR mutation in NSCLC EGFR inhibitor V600E mutation in BRAF of melanoma vemurafenib ALK translocation in NSCLC crizotinib

39
Standard Paradigm of Phase III Clinical Trials Broad eligibility Base primary analysis on ITT eligible population Don’t size for subset analysis, allocate alpha for subset analysis or trust subset analysis Only believe subset analysis if overall treatment effect is significant and interaction is significant

40
Standard Paradigm Sometimes Leads to Treating many patients with few benefiting Small average treatment effects Problematic for health care economics Inconsistency in results among studies False negative studies

41
The standard approach to designing phase III clinical trials is based on two assumptions Qualitative treatment by subset interactions are unlikely “Costs” of over-treatment are less than “costs” of under-treatment

42
Subset Analysis In the past generally used as secondary analyses Numerous subsets examined No control of type I error Trial not sized for subset analysis

43
Neither conventional approaches to subset analysis nor the broad eligibility paradigm are adequate for genomic based oncology clinical trials We need a prospective approach that includes Preserving study-wise type I error Sizing the study for the primary analysis that includes any subset analysis If there are multiple subsets, replacing subset analysis with development and internal unbiased evaluation of an indication classifier

44
Although the randomized clinical trial remains of fundamental importance for predictive genomic medicine, some of the conventional wisdom of how to design and analyze rct’s requires re-examination The concept of doing an rct of thousands of patients to answer a single question about average treatment effect for a target population presumed homogeneous with regard to the direction of treatment efficacy in many cases no longer has an adequate scientific basis

45
How can we develop new drugs in a manner more consistent with modern tumor biology and obtain reliable information about what regimens work for what kinds of patients?

46
Development is Most Efficient When the Scientific Basis for the Clinical Trial is Strong Having an important molecular target Having an important molecular target Having a drug that is deliverable at a dose and schedule that can effectively inhibit the target Having a drug that is deliverable at a dose and schedule that can effectively inhibit the target Having a pre-treatment assay that can identify the patients for whom the molecular target is driving progression of disease Having a pre-treatment assay that can identify the patients for whom the molecular target is driving progression of disease

47
When the Biology is Clear the Development Path is Straightforward Develop a classifier that identifies the patients likely (or unlikely) to benefit from the new drug Develop an analytically validated test Measures what it should accurately and reproducibly Design a focused clinical trial to evaluate effectiveness of the new treatment in test + patients
 to benefit from the new drug Develop an analytically validated test Measures what it should accurately and reproducibly Design a focused clinical trial to evaluate effectiveness of the new treatment in test + patients")
48
Using phase II data, develop predictor of response to new drug Develop Predictor of Response to New Drug Patient Predicted Responsive New Drug Control Patient Predicted Non-Responsive Off Study Targeted (Enrichment) Design
 Design")
49
Predictive Biomarkers Estrogen receptor over-expression in breast cancer Anti-estrogens, aromatase inhibitors HER2 amplification in breast cancer Trastuzumab, Lapatinib OncotypeDx gene expression recurrence score in breast cancer Low score for ER+ node - -> no chemotherapy KRAS in colorectal cancer WT KRAS = cetuximab or panitumumab EGFR mutation in NSCLC EGFR inhibitor V600E mutation in BRAF of melanoma vemurafenib ALK translocation in NSCLC crizotinib

50
Evaluating the Efficiency of Targeted Design Simon R and Maitnourim A. Evaluating the efficiency of targeted designs for randomized clinical trials. Clinical Cancer Research 10:6759-63, 2004; Correction and supplement 12:3229, 2006 Simon R and Maitnourim A. Evaluating the efficiency of targeted designs for randomized clinical trials. Clinical Cancer Research 10:6759-63, 2004; Correction and supplement 12:3229, 2006 Maitnourim A and Simon R. On the efficiency of targeted clinical trials. Statistics in Medicine 24:329-339, 2005. Maitnourim A and Simon R. On the efficiency of targeted clinical trials. Statistics in Medicine 24:329-339, 2005.

51
Relative efficiency of targeted design depends on Relative efficiency of targeted design depends on proportion of patients test positive proportion of patients test positive specificity of treatment effect for test positive patients specificity of treatment effect for test positive patients When less than half of patients are test positive and the drug has minimal benefit for test negative patients, the targeted design requires dramatically fewer randomized patients than the standard design in which the marker is not used When less than half of patients are test positive and the drug has minimal benefit for test negative patients, the targeted design requires dramatically fewer randomized patients than the standard design in which the marker is not used

52
Two Clinical Trial Designs Standard design Randomized comparison of new drug E to control C without the test for screening patients Targeted design Test patients Randomize only test + patients Treatment effect D + in test + patients Treatment effect D - in test – patients Proportion of patients test + is p + Size each design to have power 0.9 and significance level 0.05

53
RandRat = n untargeted /n targeted If D - =0, RandRat = 1/ p + 2 if p + =0.5, RandRat=4 If D - = D + /2, RandRat = 4/(p + +1) 2 if p + =0.5, RandRat=16/9=1.77
 2 if p + =0.5, RandRat=16/9=1.77")
54
Comparing T vs C on Survival or DFS 5% 2-sided Significance and 90% Power % Reduction in HazardNumber of Events Required 25%509 30%332 35%227 40%162 45%118 50%88

55
Hazard ratio 0.60 for test + patients Hazard ratio 0.60 for test + patients 40% reduction in hazard 40% reduction in hazard Hazard ratio 1.0 for test – patients Hazard ratio 1.0 for test – patients 0% reduction in hazard 0% reduction in hazard 33% of patients test positive 33% of patients test positive Hazard ratio for unselected population is Hazard ratio for unselected population is 0.33*0.60 + 0.67*1 = 0.87 0.33*0.60 + 0.67*1 = 0.87 13% reduction in hazard 13% reduction in hazard

56
To have 90% power for detecting 40% reduction in hazard within a biomarker positive subset To have 90% power for detecting 40% reduction in hazard within a biomarker positive subset Number of events within subset = 162 Number of events within subset = 162 To have 90% power for detecting 13% reduction in hazard overall To have 90% power for detecting 13% reduction in hazard overall Number of events = 2172 Number of events = 2172

57
Trastuzumab Herceptin Metastatic breast cancer 234 randomized patients per arm 90% power for 13.5% improvement in 1-year survival over 67% baseline at 2-sided.05 level If benefit were limited to the 25% test + patients, overall improvement in survival would have been 3.375% 4025 patients/arm would have been required

58
Web Based Software for Planning Clinical Trials of Treatments with a Candidate Predictive Biomarker http://brb.nci.nih.gov http://brb.nci.nih.gov

59
Regulatory Pathway for Test Companion diagnostic test with intended use of identifying patients who have disease subtype for which the drug is proven effective

60
Implications for Early Phase Studies Need to design and size early phase studies to discover an effective predictive biomarker for identifying the correct target population Need to establish an analytically validated test for measuring the predictive marker in the phase III pivotal studies

61
When the drug is specific for one target and the biology is well understood May need to evaluate several candidate tests e.g. protein expression of target or amplification of gene Phase II trials sized for adequate numbers of test positive patients and to determine appropriate cut-point of positivity

62
When the drug has several targets or the biology is not well understood Should biologically characterize tumors for all patients on phase II studies with regard to candidate targets and response moderators Phase II trials sized for evaluating candidates Opportunity for sequential and adaptive designs to improve efficiency

63
Empirical screening of expression profiles or mutations to develop predictive marker Should re-think whether to develop the drug Larger sample size required Dobbin, Zhao, Simon, Clinical Ca Res 14:108, 2008. Use of archived samples from previous negative phase III trial Use of large disease specific panel of molecularly characterized human tumor cell lines to identify predictive marker

65
“Stratification Design” “Interaction Design” Develop Predictor of Response to New Rx Predicted Non- responsive to New Rx Predicted Responsive To New Rx Control New RXControl New RX

66
Develop prospective analysis plan for evaluation of treatment effect and how it relates to biomarker Defined analysis plan that protects type I error and permits adequately powered evaluation in test + patients http://brb.nci.nih.gov Trial sized for defined analysis plan Test negative patients should be adequately protected using interim futility analysis

67
Fallback Analysis Plan Test average treatment effect at reduced level p 0 If significant claim broad effectiveness If overall effect is not significant, test treatment effect in marker + subset at level.05-p 0 If significant claim effectiveness for marker + subset Claim of significance for marker + subset should not require either Overall significance Significant interaction 67

68
Sample size for Analysis Plan To have 90% power for detecting uniform 33% reduction in overall hazard at 1% two-sided level requires 370 events. If 33% of patients are positive, then when there are 370 total events there will be approximately 123 events in positive patients 123 events provides 90% power for detecting a 45% reduction in hazard at a 4% two-sided significance level.

72
Strong confidence in test: Small r 2 and large r 1 Weak confidence in test: Small r 2 and small r 1 p 00 selected to control type I error rates

73
Bayesian Two-Stage Design RCT With Single Binary Marker 73

75
Adaptive Threshold Design Randomized clinical trial of E vs C Single candidate biomarker with K candidate cut-points Entry not restricted by biomarker value “Adaptive” in the sense that no pre-specified cut-point is provided. Eligibility is not changed during trial based on interim results 75

76
Final Analysis in Two Parts Test global null hypothesis that treatment E is equivalent to C in efficacy for all biomarker values If global null hypothesis is rejected, develop information about how effectiveness of E depends on biomarker value 76

78
Bootstrap Confidence Intervals for Threshold b

80
The confidence interval for the cut-point can be used to inform treatment decisions for future patients

83
Key Points It can be beneficial not to define a cut-point for the biomarker prior to conducting the phase III clinical trial The phase II database may be inadequate with regard to number of cases, lack of control group, different endpoint The only thing that stands in the way of a more informative phase III trial is the aspirin paradigm that the ITT analysis of the eligible population is required to serve as a basis for approval 83

84
The Biology is Often Not So Clear Cancer biology is complex and it is not always possible to have the right single predictive classifier identified with an appropriate cut-point by the time the phase 3 trial of a new drug is ready to start accrual Cancer biology is complex and it is not always possible to have the right single predictive classifier identified with an appropriate cut-point by the time the phase 3 trial of a new drug is ready to start accrual

85
85 With a Small Number of Candidate Biomarkers Biomarker Selection Design Based on Adaptive Threshold Design W Jiang, B Freidlin & R Simon JNCI 99:1036-43, 2007

86
Biomarker Selection Design Have identified K candidate biomarkers B 1, …, B K thought to be predictive of patients likely to benefit from T relative to C Have identified K candidate biomarkers B 1, …, B K thought to be predictive of patients likely to benefit from T relative to C Cut-points not necessarily established for each biomarker Cut-points not necessarily established for each biomarker Eligibility not restricted by candidate markers Eligibility not restricted by candidate markers

87
Marker Selection Design

88
Designs When there are Many Candidate Markers and Multi-marker Classifiers are of Interest

90
Adaptive Signature Design

91
The indication classifier is not a binary classifier of whether a patient has good prognosis or poor prognosis It is a “two sample classifier” of whether the prognosis of a patient on E is better than the prognosis of the patient on C

92
The indication classifier maps the vector of candidate covariates into {E,C} indicating which treatment is predicted superior for that patient The classifier need not use all the covariates but variable selection must be determined using only the training set Variable selection may be based on selecting variables with apparent interactions with treatment, with cut-off for variable selection determined by cross-validation within training set for optimal classification The indication classifier can be a probabilistic classifier 92

93
93

94
94

95
95

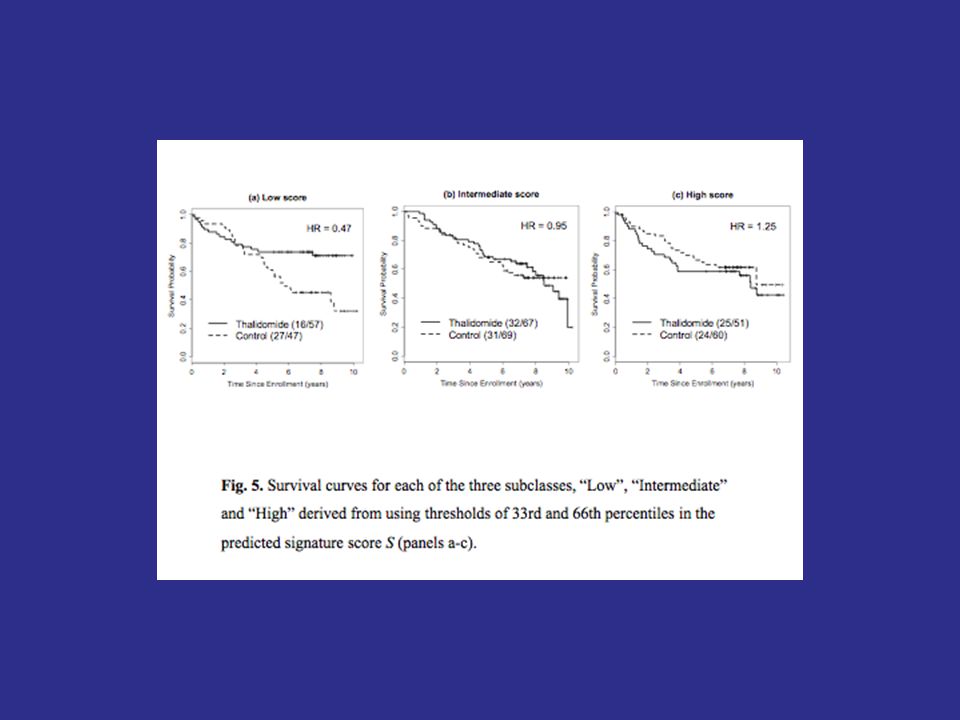
96
Treatment effect restricted to subset. 10% of patients sensitive, 400 patients. TestPower Overall.05 level test46.7 Overall.04 level test43.1 Sensitive subset.01 level test (performed only when overall.04 level test is negative) 42.2 Overall adaptive signature design85.3
 42.2 Overall adaptive signature design85.3.")
97
Overall treatment effect, no subset effect. 400 patients TestPower Overall.05 level test74.2 Overall.04 level test70.9 Sensitive subset.01 level test1.0 Overall adaptive signature design70.9

98
This approach can be used with any set of candidate predictors The approach can also be used to identify the subset of patients who don’t benefit from the new treatment when the overall ITT comparison is significant

99
Key Idea Replace multiple significance testing by development of one indication classifier and obtain unbiased estimates of the properties of that classifier if used on future patients

102
At the conclusion of the trial randomly partition the patients into K approximately equally sized sets P 1, …, P K Let D -i denote the full dataset minus data for patients in P i Omit patients in P 1 Apply the defined algorithm to analyze the data in D -1 to obtain a classifier M -1 Classify each patient j in P 1 using model M -1 Record the treatment recommendation E or C

103
Repeat the above steps for all K loops of the cross-validation (develop classifier from scratch in each loop and classify omitted patients) When cross-validation is completed, all patients have been classified once as what their optimal treatment is predicted to be
 When cross-validation is completed, all patients have been classified once as what their optimal treatment is predicted to be")
104
Let S denote the set of patients for whom treatment E is predicted optimal Compare outcomes for patients in S who actually received E to those in S who actually received C Compute Kaplan Meier curves of those receiving E and those receiving C Let z = standardized log-rank statistic

105
Test of Significance for Effectiveness of E vs C Compute statistical significance of z by randomly permuting treatment labels and repeating the entire cross-validation procedure to obtain a new set S’ and a new logrank statistic z’ Do this 1000 or more times to generate the permutation null distribution of treatment effect for the patients in S

106
The size of the E vs C treatment effect for the indicated population is (conservatively) estimated by the Kaplan Meier survival curves of E and of C in S
 estimated by the Kaplan Meier survival curves of E and of C in S")
107
Cross-Validated Adaptive Signature Design Define indication classifier development algorithm A Apply algorithm to full dataset to develop indication classifier for use in future patients M(x;A,P) Using K fold cross validation Classify patients in test sets based on classifiers developed in training sets; e.g. y i =M(x i ;A,P -i ) S={i : y i = E} Compare E to C in S and estimate size of treatment effect is an estimate of the size of the treatment effect for future patients with M(x;A,P)=E
 S={i : y i = E} Compare E to C in S and estimate size of treatment effect is an estimate of the size of the treatment effect for future patients with M(x;A,P)=E.")
108
Cross-Validated Adaptive Signature Design Approximate null distribution of Permute treatment labels Repeat complete cross-validation procedure Generate permutation distribution of the values for permuted data Test null hypothesis that the treatment effect in classifier positive patients is null using as test statistic cross-validated estimate of treatment effect in positive patients

109
70% Response to E in Sensitive Patients 25% Response to E Otherwise 25% Response to C 30% Patients Sensitive ASDCV-ASD Overall 0.05 Test 0.8300.838 Overall 0.04 Test 0.7940.808 Sensitive Subset 0.01 Test 0.3060.723 Overall Power 0.8250.918

110
25% Response to T 25% Response to C No Subset Effect ASDCV-ASD Overall 0.05 Test 0.0470.056 Overall 0.04 Test 0.040.048 Sensitive Subset 0.01 Test 0.0010 Overall Power 0.0410.048

114
The Objectives of a Phase III Clinical Trial Test the global null hypothesis that the new treatment E is uniformly ineffective relative to a control C for all patients while preserving the type I error of the study If the global null hypothesis is rejected, develop an internally validated labeling indication for informing physicians in their decisions about which patients they treat with the drug. Not a hypothesis testing problem

115
Prediction Based Clinical Trials We can evaluate our methods for analysis of clinical trials in terms of their effect on patient outcome via informaing therapeutic decision making

116
Hence, alternative methods for analyzing RCT’s can be evaluated in an unbiased manner with regard to their value to patients using the actual RCT data

118
Expected t* Year DFS Using Indication Classifier

119
Expected t* Year DFS With Conventional Analysis

120
Prediction Based Clinical Trials The resampling approach provides an internally validated way of evaluating the effectiveness of indication classifiers for informing treatment selection to improve patient outcome

121
Prediction Based Clinical Trials By switching from subset analysis to development of indication classifiers and by using re-sampling and careful prospective planning, we can more adequately evaluate new methods for analysis of clinical trials in terms of improving patient outcome by informing therapeutic decision making

122
By applying the classifier development algorithm to the full dataset D, an indication classifier is developed for informing how future patients should be treated M(x;A, D) for all x vectors. The cross validation merely serves to provide an estimate of the treatment effect for future patients with M(x;A, D) =E and to provide a significance test of the null hypothesis that the treatment effect is zero
 =E and to provide a significance test of the null hypothesis that the treatment effect is zero.")
123
The stability of the indication classifier M(x;A,D)can be evaluated by examining the consistency of classifications M(x i ;A, B) for bootstrap samples B from D.
can be evaluated by examining the consistency of classifications M(x i ;A, B) for bootstrap samples B from D.")
124
Although there may be less certainty about exactly which types of patient benefit from E relative to C, classification may be better than for standard clinical trials in which all patients are classified based on results of testing the single overall null hypothesis

125
This approach can also be used to identify the subset of patients who don’t benefit from a new regimen C in cases where E is superior to C overall at the first stage of analysis. The patients in S C = D – S are not predicted to benefit from E. Survivals of E vs C can be examined for patients in that subset and a permutation based confidence interval for the hazard ratio calculated.

127
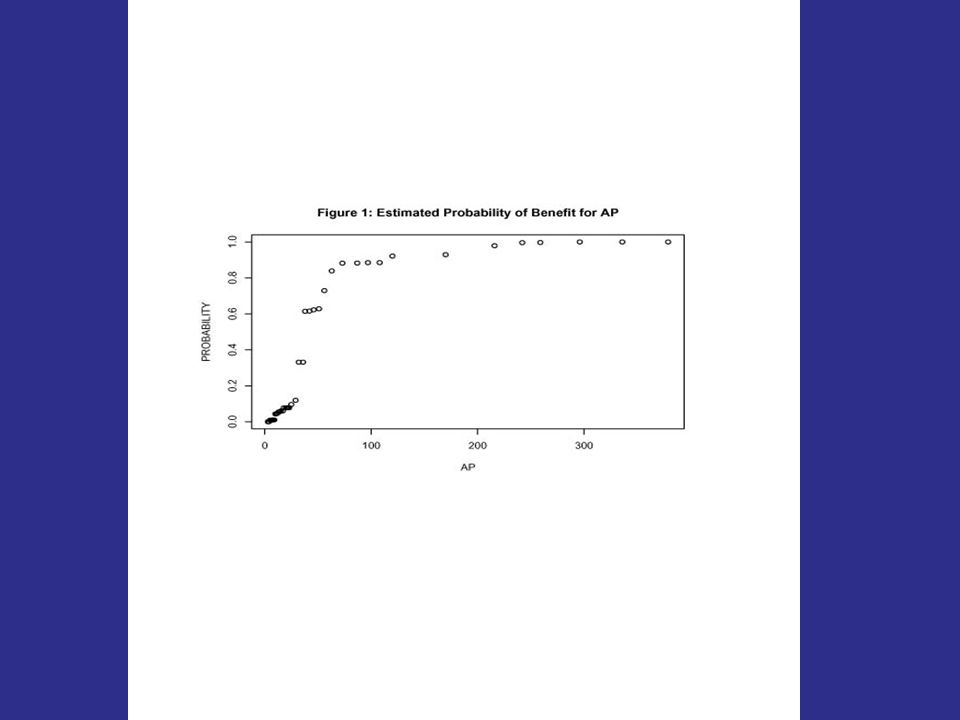
506 prostate cancer patients were randomly allocated to one of four arms: Placebo and 0.2 mg of diethylstilbestrol (DES) were combined as control arm C 1.0 mg DES, or 5.0 mg DES were combined as T. The end-point was overall survival (death from any cause). Cova Covariates: Age, performance status (pf), tumor size (sz), stage/grade index (sg), serum acid phosphatase (ap)
. Cova Covariates: Age, performance status (pf), tumor size (sz), stage/grade index (sg), serum acid phosphatase (ap).")
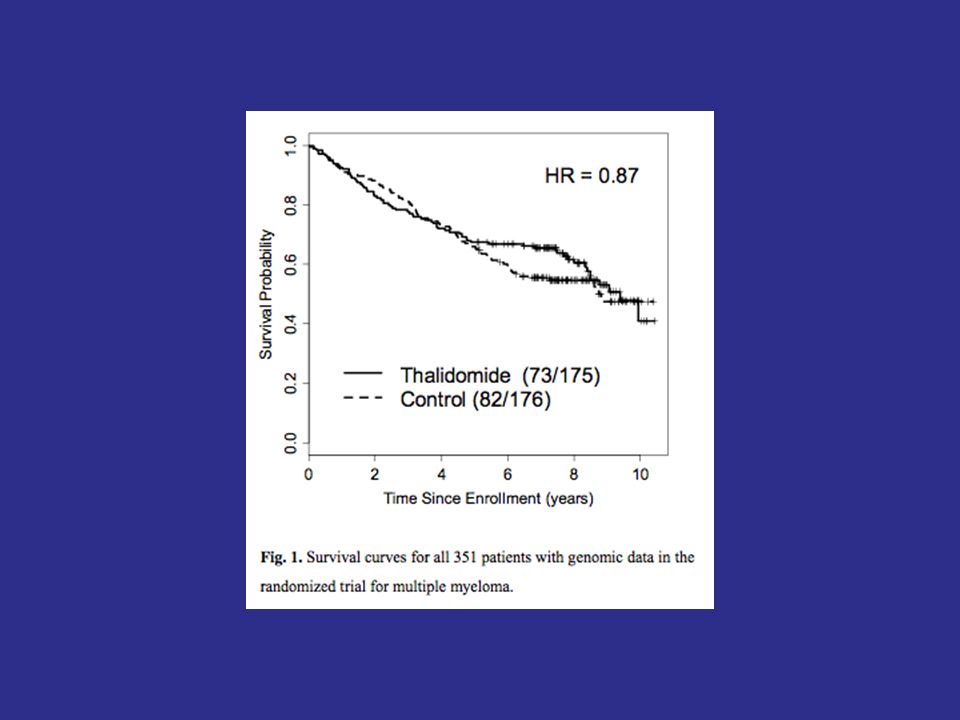
128
Figure 1: Overall analysis. The value of the log-rank statistic is 2.9 and the corresponding p-value is 0.09. The new treatment thus shows no benefit overall at the 0.05 level.

129
Figure 2: Cross-validated survival curves for patients predicted to benefit from the new treatment. log-rank statistic = 10.0, permutation p-value is.002

130
Figure 3: Survival curves for cases predicted not to benefit from the new treatment. The value of the log-rank statistic is 0.54.

132
Marker Strategy Design

133
Generally very inefficient because some (many) patients in both randomization groups receive the same treatment Often poorly informative Not measuring marker in control group means that merits of complex marker treatment strategies cannot be dissected
 patients in both randomization groups receive the same treatment Often poorly informative Not measuring marker in control group means that merits of complex marker treatment strategies cannot be dissected")
134
Validation of Predictive Biomarker Stratification Design Develop Predictor of Response to New Rx Predicted Non- responsive to New Rx Predicted Responsive To New Rx Control New RXControl New RX

135
Prospective-Retrospective Study

136
In some cases a trial with optimal structure for evaluating a new biomarker will have been previously performed and will have pre-treatment tumor specimens archived Under certain conditions, a focused analysis based on specimens from the previously conducted clinical trial can provide highly reliable evidence for the medical utility of a prognostic or predictive biomaker In some cases, it may be the only way of obtaining high level evidence

137
Prospective- Retrospectiv e Design

138
Conclusions of Simon, Paik, Hayes Claims of medical utility for prognostic and predictive biomarkers based on analysis of archived tissues can have either a high or low level of evidence depending on several key factors. These factors include the analytical validation of the assay, the nature of the study from which the specimens were archived, the number and condition of the specimens, and the development prior to assaying tissue of a focused written plan for analysis of a completely specified biomarker classifier. Studies using archived tissues from prospective clinical trials, when conducted under ideal conditions and independently confirmed can provide the highest level of evidence. Traditional analyses of prognostic or predictive factors, using non analytically validated assays on a convenience sample of tissues and conducted in an exploratory and unfocused manner provide a very low level of evidence for clinical utility.

139
Guidelines Proposed by Simon, Paik, Hayes Prospective-retrospective design 1. Adequate archived tissue from an appropriately designed phase III clinical trial must be available on a sufficiently large number of patients that the appropriate biomarker analyses have adequate statistical power and that the patients included in the evaluation are clearly representative of the patients in the trial. 2. The test should be analytically validated for use with archived tissue. 3. Testing should be performed blinded to the clinical data. 4. The analysis plan for the biomarker evaluation should be completely specified in writing prior to the performance of the biomarker assays on archived tissue and should be focused on evaluation of a single completely defined classifier. 5. The results should be validated using specimens from a similar, but separate study involving archived tissues.

140
Acknowledgements Kevin Dobbin Boris Freidlin Wenyu Jiang Aboubakar Maitournam Shigeyuki Matsui Michael Radmacher Jyothi Subramanian Yingdong Zhao

Similar presentations
























 Baselga.>")
 How.>")




 Design. Prospective Co-Development of Drugs and Companion Diagnostics 1. Develop a completely specified genomic classifier of the.>")





 Hajime Uno (Kitasato University) Tianxi Cai, Els Goetghebeur,>")

