Download presentation
Presentation is loading. Please wait.

1
Click anywhere to go on to the next slide
Tour of ViroBIKE ViroBIKE (Biological Integrated Knowledge Environment) combines: Knowledge: All completed viral genomes known to NCBI Many viral metagenomes Analytical Tools: A powerful graphical language that permits creative expression to those with no programming experience It is accessible through a virological community web site: This demonstration is best viewed as a slide show, enabling you to simulate a session and make changes in cursor position more obvious. To do this, click Slide Show on the top tool bar, then View show. Click anywhere to go on to the next slide
 combines: Knowledge: All completed viral genomes known to NCBI Many viral metagenomes. Analytical Tools: A powerful graphical language that permits creative expression to those with no programming experience. It is accessible through a virological community web site: This demonstration is best viewed as a slide show, enabling you to simulate a session and make changes in cursor position more obvious. To do this, click Slide Show on the top tool bar, then View show. Click anywhere to go on to the next slide.")
2
Tour of ViroBIKE In this tour, you'll see how to: Log onto ViroBIKE
Speak BioBIKE (the language of ViroBIKE) Identify and define a group of viruses Analyze a group of viruses Analyze the upstream regions of a group of genes Collect sequences and generate FastA files Compare sequences to a database (Blast) Analyze Blast results Do multiple sequence alignment Make phylogenetic trees Construct a novel tool: (example) Analysis of palindromes
 Identify and define a group of viruses. Analyze a group of viruses. Analyze the upstream regions of a group of genes. Collect sequences and generate FastA files. Compare sequences to a database (Blast) Analyze Blast results. Do multiple sequence alignment. Make phylogenetic trees. Construct a novel tool: (example) Analysis of palindromes.")
3
Access this site at htpp://biobike.csbc.vcu.edu
To get to ViroBIKE, click a link to one of the public sites/ To see more tours like this one, click Guided tours of BioBIKE Access this site at htpp://biobike.csbc.vcu.edu

4
Your name (no spaces) - Enter anything you like as a login name, but no spaces or symbols. - address is optional but may be useful if you want to send in questions or complaints. - Click New Login

5
Function palette Workspace The BioBIKE environment is divided into three areas as shown. You'll bring functions down from the function palette to the workspace, execute them, and note the results in the results window Results window

6
Two very important buttons on the function palette:
On-line help (general) Something went wrong? Tell us! HELP! PROBLEM
 Something went wrong Tell us! HELP! PROBLEM.")
7
Our Story Suppose you want to analyze sequences from mycobacterial viruses, particularly the virus called pipefish. The first step is to define the set of mycobacterial viruses. Click the DEFINITION button on the function palette.

8
Clicking on any palette button brings down a function or data into the workspace.
Click DEFINE.

9
A DEFINE box is now in the workspace.
Before continuing with the problem, let's consider what function boxes mean.

10
General Syntax of BioBIKE
Function-name Argument (object) Keyword object Flag The basic unit of BioBIKE is the function box. It consists of the name of a function, perhaps one or more required arguments, and optional keywords and flags. A function may be thought of as a black box: you feed it information, it produces a product.
 Keyword. object. Flag. The basic unit of BioBIKE is the function box. It consists of the name of a function, perhaps one or more required arguments, and optional keywords and flags. A function may be thought of as a black box: you feed it information, it produces a product.")
11
General Syntax of BioBIKE
Function-name Argument (object) Keyword object Flag SIN angle A function you’re already familiar with is the Sin function. You feed it an angle, it produces the sin of the angle. In BioBIKE, you provide information by clicking on a gray input box to open it up for entry.
 Keyword. object. Flag. SIN. angle. A function you’re already familiar with is the Sin function. You feed it an angle, it produces the sin of the angle. In BioBIKE, you provide information by clicking on a gray input box to open it up for entry.")
12
General Syntax of BioBIKE
Function-name Argument (object) Keyword object Flag SIN 30 A box that is white and outlined in read is open for entry. Type into it an appropriate value, then close it by pressing Enter or Tab. All input boxes must be closed before a function may be executed.
 Keyword. object. Flag. SIN. 30. A box that is white and outlined in read is open for entry. Type into it an appropriate value, then close it by pressing Enter or Tab. All input boxes must be closed before a function may be executed.")
13
General Syntax of BioBIKE
Function-name Argument (object) Keyword object Flag SIN 30 But wait! Is that 30 degrees or 30 radians? You modify how the function works by choosing options from the options menu accessible through the options icon.
 Keyword. object. Flag. SIN. 30. But wait! Is that 30 degrees or 30 radians You modify how the function works by choosing options from the options menu accessible through the options icon.")
14
General Syntax of BioBIKE
Function-name Argument (object) Keyword object Flag SIN 30 DEGREES (Suppose you selected the DEGREES flag). But wait again! Maybe you're working with an angle that has been rotated some number of degrees. You can modify the function further by returning to the options menu.
 Keyword. object. Flag. SIN. 30. DEGREES. (Suppose you selected the DEGREES flag). But wait again! Maybe you re working with an angle that has been rotated some number of degrees. You can modify the function further by returning to the options menu.")
15
General Syntax of BioBIKE
Function-name Argument (object) Keyword object Flag OFFSET angle SIN 30 DEGREES Functions, options… Let's summarize:
 Keyword. object. Flag. OFFSET. angle. SIN. 30. DEGREES. Functions, options… Let s summarize:")
16
General Syntax of BioBIKE
Function-name Argument (object) Keyword object Flag Function boxes contain the following elements: Function-name (e.g. SEQUENCE-OF or LENGTH-OF) Argument: Required, acted on by function Keyword clause: Optional, more information Flag: Optional, more (yes/no) information
 Keyword. object. Flag. Function boxes contain the following elements: Function-name (e.g. SEQUENCE-OF or LENGTH-OF) Argument: Required, acted on by function. Keyword clause: Optional, more information. Flag: Optional, more (yes/no) information.")
17
General Syntax of BioBIKE
Function-name Argument (object) Keyword object Flag … and icons to help you work with functions: Option icon: Brings up a menu of keywords and flags Action icon: Brings up a menu enabling you to execute a function, copy and paste, information, get help, etc Clear/Delete icon: Removes information you entered or removes box entirely
 Keyword. object. Flag. … and icons to help you work with functions: Option icon: Brings up a menu of keywords and flags. Action icon: Brings up a menu enabling you to execute a function, copy and paste, information, get help, etc. Clear/Delete icon: Removes information you entered or removes box entirely.")
18
Back to our story… we were defining the set of sequenced mycobacterial phages.
The DEFINE function has two arguments: the name of the variable being defined and its value. Click on the argument marked variable to provide the name.

19
That opens the input box for entry
That opens the input box for entry. Type in a reasonable name for the set – anything you like, but no spaces. Then remember to press Enter or Tab to close the input box. Tab differs from Entry in that it automatically brings you to the next input box.

20
Now provide the values for this set.
How to do that? Our strategy: Choose all viruses from the set of known viruses that have "mycobact" in their names. To do this, go to the GENOME button in the paletten and click on ORGANISM-NAMED.

21
Next you select the input box, type in "mycobact", and press Enter.

22
Of course, there is no virus named "mycobact"
Of course, there is no virus named "mycobact". You want viruses with those letters as part of the name. So mouse over the options icon and click on the IN-PART flag.

23
Now the definition is ready for execution
Now the definition is ready for execution. Mouse over the action icon of the DEFINE box and click on Execute. The action icon governs only what is within the box. If you had clicked Execute within the action icon of the orange ORGANISM-NAMED box, you would have been given the viruses you're looking for, but the definition would not have taken place.

24
Executing the function produces a list of viruses in the Result Window.
The Result Window contains results of functions in a form that can be used for further manipulations. If you want a list that is easy for humans to read, you'd do something different. How many mycobacterial phages are there in this list? Click on the LISTS-TABLES button to find an appropriate function.

25
Menus are arranged hierarchically, to facilitate finding unknown functions. However, if you know what you're looking for, you can always get the function from the alphabetized ALL button. Click on COUNT-OF to get the count of the list of phages (LENGTH-OF would work equally well).
.")
26
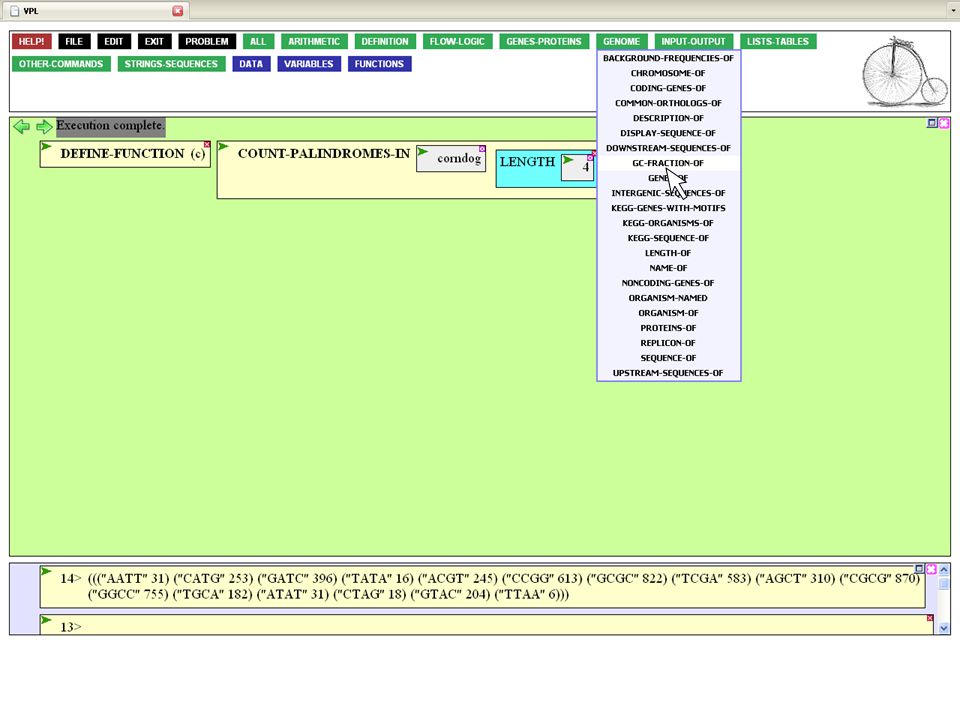
You defined the list of phages as mycobacterial-phages
You defined the list of phages as mycobacterial-phages. You could type this into the entry box, but there's an easier (and more reliable) way. Anything you define becomes part of your personal language and can be accessed from the VARIABLES button. Click on the name of your list
 way. Anything you define becomes part of your personal language and can be accessed from the VARIABLES button. Click on the name of your list.")
27
The name of the list is brought down into the input box
The name of the list is brought down into the input box. Now execute the completed function.

28
There are 28 mycobacterial phages known to ViroBIKE.
How long are the phages? Back to the LIST-TABLES button. (If you guessed that GENOME was the correct button in which to find a LENGTH-OF function, you'd also have been right)
")
29
Click on LENGTHS-OF to give you the lengths of each phage within the list. (LENGTH-OF would give you the length of the list itself)
.")
30
Executing the function once you complete it gives you a list of lengths in the Results Window, corresponding to the list of phages. The first phage has a length of 78441, the second, 52797, etc. What is the average length? That's a statistical question, so go to the ARITHMETIC button

31
Click on MEAN from amongst the statistical functions.

32
Mean of what? One easy way to provide the argument is to cut and paste the function you already constructed. Cut and paste operations are found through the action icon.

33
Cutting puts on the BioBIKE clipboard everything in the box governed by the action menu.
Now paste the box into the input box of MEAN.

34
Executing this function gives you the desired answer: the average length of the mycobacterial phages is about 70,000 nucleotides. Now we turn to a specific mycobacterial phage, called pipefish. What genes does it have in its genome? Find an appropriate function on the GENOME button.

35
GENES-OF certainly looks plausible.

36
ViroBIKE knows each virus through its GenBank accession number (e. g
ViroBIKE knows each virus through its GenBank accession number (e.g. NC_008199) and also through a nickname (e.g. pipefish). You can type either one into the input box of GENES-OF. Executing the function produces a list of the phage's genes. How long are the gene's upstream regions?
 and also through a nickname (e.g. pipefish). You can type either one into the input box of GENES-OF. Executing the function produces a list of the phage s genes. How long are the gene s upstream regions")
37
How long are the sequences upstream from Pipefish's genes
How long are the sequences upstream from Pipefish's genes? You can get the sequence upstream from a specific gene or the sequences upstream from a group of genes through the SEQUENCE-UPSTREAM-OF function.

38
Genes can be referred to by their full names (e. g. NC_008199
Genes can be referred to by their full names (e.g. NC_ Pipefishp1) or by their nicknames (e.g. Pipefishp1). Entering a nickname into the entry box and executing the function gives the upstream sequence. What about upstream sequences of all Pipefish genes? Clear the entry box (using the clear icon) and try again.
 or by their nicknames (e.g. Pipefishp1). Entering a nickname into the entry box and executing the function gives the upstream sequence. What about upstream sequences of all Pipefish genes Clear the entry box (using the clear icon) and try again.")
39
We're going to cut and paste the set of genes of Pipefish into the entry box of SEQUENCE-UPSTREAM-OF. Of course, we could have used DEFINE to give a name to the genes and refered to them by that name.

40
Now paste the function on the BioBIKE clipboard into the entry box.

41
We could now execute the SEQUENCE-UPSTREAM-OF function, but that would just give us several dozen sequences. We want their lengths, which we can get by putting this function in the input box of the LENGTHS-OF function, as we did before. This could be done by cutting and pasting, but there's a simpler way…

42
Marking a box by clicking Surround with puts it into the first argument of the next function you select. Click on LENGTHS-OF.

43
Executing the completed function gives a list of upstream sequences, many of them zero. Only the first 100 are shown, but all are in memory, available for further computation. That's Pipefish. What about all mycobacterial phages? First let's define their genes.

44
Note that GENES-OF mycobacterial-phages gives a list of lists (see right). This is sometimes useful, but in this case we want them all thrown into one bag. We want to simplify the list. 9> ((Wildcat genes) (Corndog genes) (Pipefish genes)…)
 (Corndog genes) (Pipefish genes)…)")
45
Go to the action menu of GENES-OF and mark the function for surrounding, then get the SIMPLIFY-LIST function.

46
Now the list is a simple list of genes (see right).
With myco-genes suitably defined, you can modify an earlier function to act on this new set. 10> (Wildcat genes… Corndog genes… Pipefish genes…)
")
47
The results now give the lengths of well over a thousand mycobacterial genes. A frequency graph will help us understand these numbers. First we need to bin the data.

48
I chose to bin data every 10 nucleotides, for intergenic sequences of lengths 0 to 500 nucleotides. Note that the biggest category by far are genes in the 0-10 nt bin. We'll export the results and use Excel to make the graph.

49
The strategy is to write the data to your disk space on the ViroBIKE server and then download it to your own computer.

50
We have to specify what we want to write to disk and the name of the file we want to write to. The what is the result we just got.

51
Here we give as the argument the PREVIOUS RESULT
Here we give as the argument the PREVIOUS RESULT. Of course it would have been possible to cut/paste the previous function, but this way allows us to avoid re-executing BIN-DATA-OF.

52
Excel likes tab-delimited files (TABBED is synonymous with TAB-DELIMITED).
.")
53
After executing WRITE, we'd like to find and download the file we've just written

54
I can click on the file, download it, and…

55
…import it directly into Excel (or whatever) to make nice graph.
 to make nice graph.")
56
More to do,… not enough space.
Clear the screen with the clear icon of the Workspace.

57
Now to identify to a first approximation the protein families occupied by Pipefish proteins.
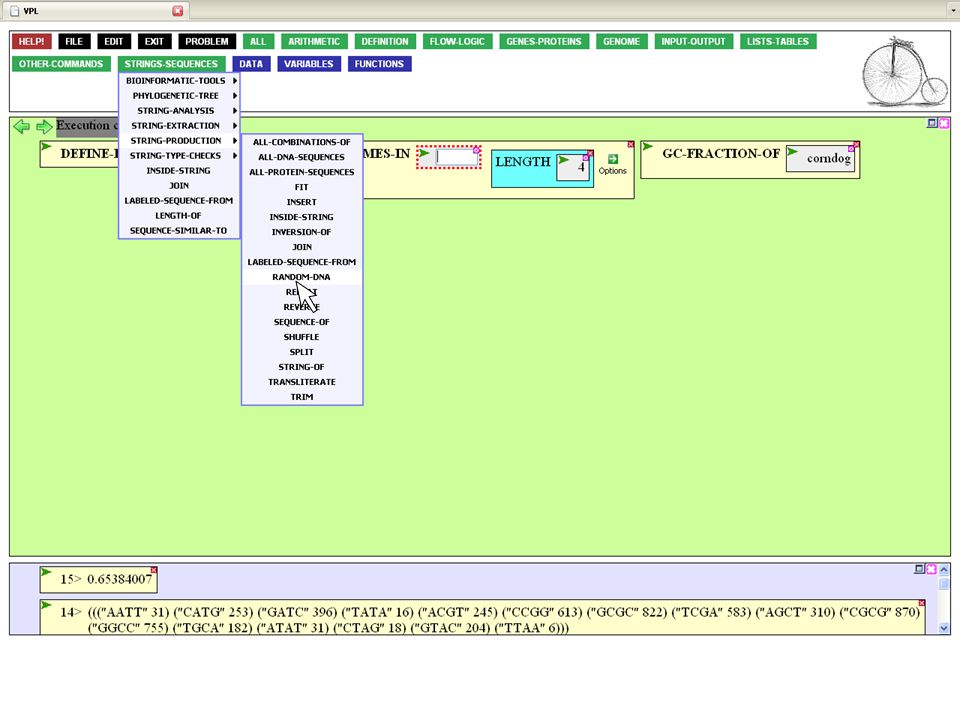
We can approach this problem by asking for each Pipefish protein what viral proteins share sequence similarity with it. Sequence similarity functions would be found in the STRINGS-SEQUENCES menu.

58
SEQUENCE-SIMILAR-TO to allows you access to different sequence search algorithms. The most commonly used is Blast, but you can also search by the number of sequence mismatches or by sequence pattern.

59
We could consider as the query each Pipefish protein one at a time, but just for fun, let's try the whole batch at once. Click on the first argument to SEQUENCE-SIMILAR-TO and then…

60
… go to the GENOME menu to get a function that will return all proteins of Pipefish.

61
After typing in Pipefish into the input box of PROTEINS-OF, select the target input box.
What to put in it? We could search over all mycobacterial phage protein, over all bacteriophage protein,… Go to the DATA menu and make your choice.

62
I choose all known viruses.

63
It's ambiguous whether we intend to compare Pipefish proteins to the annotated proteins of the known viruses (BlastP) or to a virtual translation of the viruses' DNA (TBlastN). Click on the options icon and then PROTEIN-VS-PROTEIN to disambiguate the function.

64
SEQUENCE-SIMILAR-TO produces both a result (a table), which can be manipulated further, and a display of the result, which is just for human consumption. The table is organized the same way as the display: by row (1 through …) and column ("QUERY", "Q-START" etc).
 and column ( QUERY , Q-START etc).")
65
So far most of things we did have been relatively straightforward
So far most of things we did have been relatively straightforward. The difficult thing was knowing what function to use. From here on, things will be a wee bit different. We'll be doing operations that are generally considered the province of computer geeks – loops, mapping,… never mind. After acclimating yourself to BioBIKE, what follows may grow to be more do-able, even if you've never previously touched a computer language. In the meantime, you can probably get the gist of what is being done. But if you want to stop here, no shame.

66
The Blast search gave us many hundreds of hits
The Blast search gave us many hundreds of hits. How do we make sense of the results? One thing we can do is to count how many times each Pipefish protein found similar proteins in other viruses. Which proteins are part of large families? The first step is to give the Blast results a name, using DEFINE.

67
Our strategy is to go through each line of the table, from 1 to the end. From each line, we extract the query (the Pipefish protein) and use it to construct a new table. The table will consist of the Pipefish proteins and the number of times each appears as a query in the Blast results. This strategy requires constructing a logical series of instructions to be repeated for each line.

68
The key instruction in the series is to update a table (I called it protein-count), adding one to the count of a specific protein each time that protein appears in the Blast results. Before I get to this instruction, I need to describe how to obtain the name of the protein.

69
Representation of pipefish-blast table
I can get the full protein name from the table containing the blast result, looking in a specific line under the column labeled "QUERY". But the full name (e.g. NC_ p-pipefishp4) is rather bulky for a row header in the new table. I'd rather use the nickname. I get that using the NAME-OF function.
 is rather bulky for a row header in the new table. I d rather use the nickname. I get that using the NAME-OF function.")
70
Having figured out what I want to do with each line of the result (extract the name, update the the protein-count table), I now have to explain where to get the line numbers to which the instructions will be applied. The first dimension of the pipefish-blast table is its rows, and the labels of the rows are the numbers 1 to the number of the last line. I consider each of these numbers one at a time.

71
I've now constructed protein-count, the table of counts for each protein of Pipefish. I display the table, specifying how wide to make each column and specifying that the table should be inverted (the headers applying to rows rather than columns). Executing this function gives me – finally – the result I'm after.
. Executing this function gives me – finally – the result I m after..")
72
I now know for each Pipefish protein how many similar proteins there are amongst all viruses. Pipefishp28 is a protein with lots of matches. I'd like to examine why this protein (or parts of it) is so popular, focusing on members of its family in mycobacterial phages.
 is so popular, focusing on members of its family in mycobacterial phages..")
73
First, I repeat the Blast I did before, but using a single protein, pipefishp28, as the query and mycobacteria as the target. The result, as before, is a table, with each numbered row consisting of information about a particular match. Note that a target protein may arise multiple times if regions of similarity are found more than once in the protein.

74
I want to extract from the table the proteins found by Blast so I can align their sequences. I could get the proteins through a loop similar to the one shown a few slides ago, but there is another way – mapping – that is sometimes more convenient. The first step is to construct a function that gets me the desired protein from one line of the table. Rather than give a name to the table, I refer to it as the previous result.

75
That gives me the name of the protein, rather than the protein itself, i.e. the information structure from which I can obtain sequence and other information. So I need to convert the name to the protein, using the PROTEIN-NAMED function.

76
Now I apply that function to each of the 46 lines of the blast result, giving me a list of proteins found in mycobacterial phages similar to pipefishp28.

77
Finally, I align the new previous result (i.e., the list of proteins). Acting sequentially on previous results allows me to quickly move from step to step. The alignment is given as a result…

78
… and also displayed as the output from the alignment program Clustal.

79
I can go further, constructing from the alignment a phylogenetic tree (BioBIKE uses Phylip). This is a new capability to BioBIKE, and while the menu structure greatly facilitates the navigation through Phylip's great number of options, we still need to build a simple, TREE-OF function that automates the process, using default choices and offering a few key variants as options. The functions return a computer-readable tree for further analysis and also…

80
… displays the tree graphically.

81
Thus far, we've used primarily existing tools (Blast, Clustal, Phylip), bringing them into a common interface and integrating them with the ViroBIKE database. There are many instances, however, where no existing tool suffices, and it is necessary to build one from scratch (or at least from what BioBIKE provides as scratch). What follows is an example of a relatively novel problem requiring tools made by hand.
. What follows is an example of a relatively novel problem requiring tools made by hand.")
82
Bacteriophage are at war with bacteria
Bacteriophage are at war with bacteria. Bacteria throw restriction enzymes at phage, and phage respond in several ways. One way is by minimizing the targets for those restriction enzymes. Since restriction enzyme targets are often palindromic sequences, it becomes of some interest to examine the frequencies of palindromes in mycobacterial phage sequences.

83
First, I teach BioBIKE what a palindrome is, focusing for now on palindromes of six nucleotides, without gaps. Such a palindrome is three nucleotides, abc, followed by its inversion, c'b'a', where a' is the complement of a. For a specific triplet, I can define the palindrome by joining the triplet to its inversion.

84
Then I apply this function to all 64 possible triplets, and give the resulting set of palindromes a name, palindrome6.

85
COUNTS-OF is used to count all the hexanucleotide palindromes in Pipefish, and the result is displayed so that it is easier to read and easier to save. Now that I've figured out how to do this, I can packaged the knowledge so I don't have to go through this exercise again, by defining a new function.

86
Defining this function is mostly a matter of cutting and pasting what I did in the previous slides.
Once the new function is defined, it becomes part of my language, appearing in the FUNCTION menu. Before using it, I'll compress the function definition.

87
While no substitute for a 24" screen, compressing large functions and expanding them when needed makes life much easier.

88
Now I use my new function, trying it out on a different mycobacterial phage, with a different sized palindrome.

89
All the palindromes that are AT-rich have low numbers
All the palindromes that are AT-rich have low numbers. Have I discovered a wondrous new phenomenon? Or does this phage simply have a very high GC-content? It's the work of a moment to find out. First, what is the GC-content of corndog?

91
Corndog turns out to be 65. 4% GC
Corndog turns out to be 65.4% GC. That's high, but does it explain the counts in AT-rich palindromes? Some combinatorics would help here,… but simulation is quicker: simply replace corndog with random DNA of the same base composition.

93
The randomized sequence has low counts for AT-rich palindromes, but not nearly so low as the real corndog genome. There are cleverer ways of making random sequences for comparison (e.g. maintaining codon usage), but this is a good first indication of bias against the palindromic sequences. CTAG is particularly biased.
, but this is a good first indication of bias against the palindromic sequences. CTAG is particularly biased.")
94
ViroBIKE You've seen a knowledge environment in which:
Knowledge and tools are integrated. Data conversion is seldom necessary. The language is uniform, facilitating access to many popular tools through a common interface. The language is as flexible as any general purpose language, permitting construction of new tools. The programming language is easy to pick up, using graphical conventions familiar to those who don't program. The environment is well suited for teaching the concepts of molecular biology through computational experiment.

95
Collaborators Michael Chaplin Johnny Casey (Sequoia Cons.) Sarah Cousins (now VaTech) Michiko Kato (now UC Davis) Hailan Liu JP Massar (Berkeley) James Mastros (now Philip Morris) Bogdan Mihai John Myers (Sequoia Cons.) Nihar Sheth Jeff Shrager (Carnegie Inst.) Arnaud Taton Hien Truong Andy Whittam (Washington & Jefferson) Ena Xiao … and many participating students Development of BioBIKE was funded by a grant from the National Science Foundation Contact Jeff Elhai, Center for the Study of Biological Complexity, Virginia Commonwealth University ( ) (Tel)
 Sarah Cousins (now VaTech) Michiko Kato (now UC Davis) Hailan Liu JP Massar (Berkeley) James Mastros (now Philip Morris) Bogdan Mihai John Myers (Sequoia Cons.) Nihar Sheth Jeff Shrager (Carnegie Inst.) Arnaud Taton Hien Truong Andy Whittam (Washington & Jefferson) Ena Xiao … and many participating students. Development of BioBIKE was funded by a grant from the National Science Foundation Contact Jeff Elhai, Center for the Study of Biological Complexity, Virginia Commonwealth University ( ) (Tel)")
Similar presentations











, rating INT, age REAL) Maintenances (sin INT, planeId INT, day.>")



 ASSIGNMENT 2.>")



