Download presentation
Presentation is loading. Please wait.

1
Good Laboratory Practice (GLP)
Angela Ng Min Hwei, PhD Tissue Engineering Centre Faculty of Medicine, UKM

2
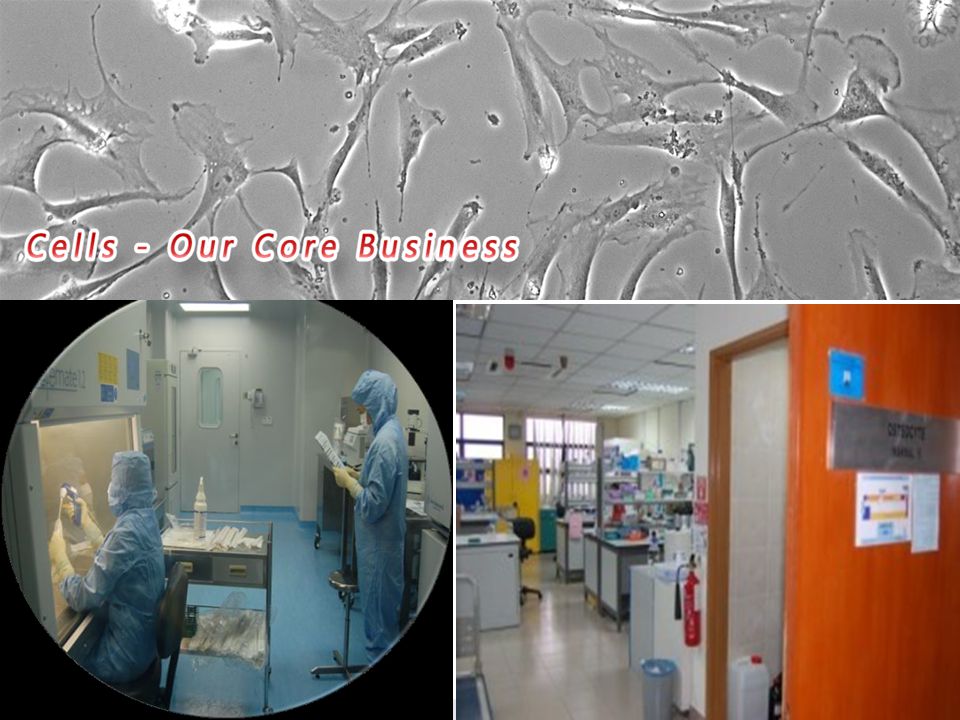
Tissue Engineering Centre (TEC)
Established in 2008 as Centre of Research Excellence in UKMMC Completed building of a GMP facility for cell and tissue processing in 2011 Obtained certificate of compliance to GMP standards in 2012 Currently, running clinical trial for MyDerm™, a tissue engineered skin R&D facility has completed ISO9001 and in the midst of ISO audits

4
Medical Product Development Process
*Time line approximately yrs Non-clinical/ Pre-clinical trial Clinical TrialPhase I Discovery Safety of personnel & facilities Safety & toxicology in in vitro and animal studies Safety in human (normal subjects) CT Phase II CT Phase III (Multi-centre) Marketing CT Phase IV Post-marketing Efficacy in patients Continuous monitoring of adverse effects
 CT Phase II. CT Phase III. (Multi-centre) Marketing. CT Phase IV. Post-marketing. Efficacy in patients. Continuous monitoring of adverse effects.")
5
Role of Laboratories in Medical Product Development Process
Research Laboratories Discovery & development of new drugs / device/therapy Fundamental research/mechanisms of diseases Animal Laboratories In vivo studies Animal models / preclinical studies Safety and sensitivity testing Calibration Laboratories Equipment & device calibration

6
Clinical Testing & Analytical Laboratories
Non clinical Testing & Analytical Laboratories Toxicology Mutagenicity Safety pharmacology Bioequivalence / bioavailability pharmacokinetics Clinical Testing & Analytical Laboratories Screening & Diagnosis (Enrolment : inclusion criteria exclusion) Quality control Trial monitoring : Verify effects of drugs clinical efficacy Monitoring of adverse effects safety Data analysis Verification Manufacturing Laboratories Production of drugs, cell-based therapy, plasma products, medical device
 Quality control. Trial monitoring : Verify effects of drugs clinical efficacy. Monitoring of adverse effects safety. Data analysis. Verification. Manufacturing Laboratories. Production of drugs, cell-based therapy, plasma products, medical device.")
7
Why do we need STANDARDS?
Laboratories are required to be Accredited or Compliant to a set of defined STANDARDS Standards are documented agreements containing technical specifications or other precise criteria used consistently as rules, guidelines and definitions of characteristics to ensure that material, products, process and services are fit for its claimed purpose.

8
Standards required at various stages of medical product development
Good practice in Lab/ ISO / HIRAC GLP GCP GMP / GTP GLCP Non-clinical/ Pre-clinical trial Clinical Trial Phase I (Safety in human, tolerance of test drugs, define human pharmacokinetics) Discovery Safety of personnel & facilities ISO (International Organisation of Standardisation) is a worldwide federation of national standards from 130 countries NGO established in 1947 with a mission to promote development of standardisation and related activities in the world with a view to : - facilitate international exchange of goods - develop coorperation in spheres of intellectual, scientific, technological and economic activity GUIDELINES FOR HAZARD IDENTIFICATION, RISK ASSESSMENT AND RISK CONTROL (HIRARC), 2008 by DOSH (Department of Occupational Safety & Health) Safety & toxicology in in vitro and animal studies GCP is the basis for quality standards, ethical conduct and regulatory compliance GDP CT Phase II (Efficacy, dose-effect relationship) CT Phase III (full-scale, often multi-centre clinical efficacy trials in patients) Clinical trial data submit for approval CT Phase IV Post-marketing
 Discovery. Safety of personnel & facilities. ISO (International Organisation of Standardisation) is a worldwide federation of national standards from 130 countries. NGO established in 1947 with a mission to promote development of standardisation and related activities in the world with a view to : - facilitate international exchange of goods. - develop coorperation in spheres of intellectual, scientific, technological and economic activity. GUIDELINES FOR HAZARD IDENTIFICATION, RISK ASSESSMENT AND RISK CONTROL (HIRARC), 2008 by DOSH (Department of Occupational Safety & Health) Safety & toxicology in in vitro and animal studies. GCP is the basis for quality standards, ethical conduct and regulatory compliance. GDP. CT Phase II. (Efficacy, dose-effect relationship) CT Phase III. (full-scale, often multi-centre clinical efficacy trials in patients) Clinical trial data submit for approval. CT Phase IV. Post-marketing.")
9
Standards available International Organisation of Standardisation (ISO) Good Clinical Practice (GCP) Good Manufacturing Practice (GMP) Good Distribution Practice (GDP) Good Tissue Practice (GTP) Good Clinical Laboratory Practice (GLCP) OECD Principle of Good Laboratory Practice (OECD GLP)
 Good Distribution Practice (GDP) Good Tissue Practice (GTP) Good Clinical Laboratory Practice (GLCP) OECD Principle of Good Laboratory Practice (OECD GLP)")
10
Good Clinical Practice (ICH-GCP)
Standards must be adhered during Clinical Drugs Trial for design, conduct, performance, monitoring, auditing, recording, analysis and reporting in order to provide assurance that data and reported results are credible and accurate and integrity and confidentiality of trial subjects are protected Section 2.13 Systems with procedure that assure the quality of every aspect of the trial should be implemented

11
ISO Standards for laboratories
For Accreditation of Competence of Testing and Calibration Laboratories ISO/IEC Guide 25 (1990) – replaced by ISO/IEC 17025 ISO/IEC (2005) new standards which meets those of ISO 9001 and covering both technical and management requirements. In general, more mandatory in nature compared to the recommendations if ISO 25 ISO/IEC (2002) - expanded, requirements for quality and competence of Medical Laboratories e.g. JPMD, UMBI
 – replaced by ISO/IEC ISO/IEC (2005) new standards which meets those of ISO 9001 and 9002 covering both technical and management requirements. In general, more mandatory in nature compared to the recommendations if ISO 25. ISO/IEC (2002) - expanded, requirements for quality and competence of Medical Laboratories e.g. JPMD, UMBI.")
12
Good Manufacturing Practice (WHO-GMP)
A standard that should be followed by manufacturers of registered pharmaceutical / traditional products and cosmetics to ensure that the product manufactured is safe, efficacious and of quality. Satisfactory GMP compliance is one of the requirements for product registration, as well as to apply for a manufacturing license with the Drug Control Authority (DCA). Uncontrolled manufacturing operations may be detrimental to consumer health.
. Uncontrolled manufacturing operations may be detrimental to consumer health.")
13
Good Tissue Practice (GTP )
Guideline developed by FDA specifically to establishments that manufacture Human Cell and Tissue Products (HCT/Ps). Not implemented in M’sia yet. Similar to GMP but taking into considerations that the nature of the product is very different (biologics vs drugs) Additional guidelines e.g.: All donor eligibility requirements Prevention of the introduction, transmission, or spread of communicable diseases, Ship in quarantine
. Not implemented in M’sia yet. Similar to GMP but taking into considerations that the nature of the product is very different (biologics vs drugs) Additional guidelines e.g.: All donor eligibility requirements. Prevention of the introduction, transmission, or spread of communicable diseases, Ship in quarantine.")
14
Good Distribution Practice (GDP)
Defined as important steps that should be considered in the storage, transportation and distribution (cold chain, integrity of labels & everyone involved in the distribution channel, etc) of registered products / notified cosmetics, including associated materials in order to preserve its characteristics and quality until it reaches the consumer. Generally, GDP is included into the scope of a GMP inspection.
 of registered products / notified cosmetics, including associated materials in order to preserve its characteristics and quality until it reaches the consumer. Generally, GDP is included into the scope of a GMP inspection.")
15
Good Clinical Laboratory Practice (WHO- GCLP)
Applies those principles established under GLP for data generation used in regulatory submissions relevant to the analysis of samples from a clinical trial. Ensures the reliability and integrity of data generated by analytical laboratories. Ensures that the objectives of the GCP principles are carried out.

16
Good Laboratory Practice (OECD GLP)
GLP applies to nonclinical studies conducted for the assessment of the safety or efficacy of chemicals (including pharmaceuticals). GLP helps assure regulatory authorities that the data submitted are a true reflection of the results obtained during the study and can therefore be relied upon when making risk/safety assessments.
. GLP helps assure regulatory authorities that the data submitted are a true reflection of the results obtained during the study and can therefore be relied upon when making risk/safety assessments.")
17
OECD Principles of GLP Define and describe a quality system concerned with the organisational processes and conditions under which a non-clinical health and environmental safety study is conducted Non-clinical laboratory study means in vivo (in experimental animals) or in vitro experiments in which test articles are studied prospectively in test systems under laboratory conditions to determine their safety
 or in vitro experiments in which test articles are studied prospectively in test systems under laboratory conditions to determine their safety.")
18
Origin of Good laboratory Practice (GLP)
1st evolved in USA, 1970s as a result of concerns about validity of preclinical safety data submitted to FDA for new drug applications 1950s s : 40% toxicology testing were carried out by Industrial Biotest Laboratories (IBT) , FDA imposed requirements for manufacturers to prove drug effectiveness and responsible in determining if benefits outweigh risks
 , FDA imposed requirements for manufacturers to prove drug effectiveness and responsible in determining if benefits outweigh risks.")
19
History of GLP GLP was first introduced in New Zealand and Denmark in 1972. Most notably, the lab that ran tests for big companies such as Procter and Gamble called Industrial Bio Test. It was discovered that mice that they had used to test lotion and deodorants had developed cancer and died. 867 audits of IBT performed by FDA (1962 Law of Drug Amendments) : 518 were found to be invalid due to numerous discrepancies between study and data or were so poor that police investigators could not piece together what work had been done...even though IBT superficially delivered the test results their contracts with the manufacturers specified.
 : 518 were found to be invalid due to numerous discrepancies between study and data. or were so poor that police investigators could not piece together what work had been done...even though IBT superficially delivered the test results their contracts with the manufacturers specified.")
20
Industrial Bio Test lab threw the dead mice and covered results deeming the products good for human use. FDA found 4 IBT managers guilty of frauds Those involved in production, distribution and sales for the IBT lab eventually served jail time. GLP was instituted in US following these cases of fraud generated by toxicology labs in data submitted to the FDA by pharmaceutical companies.

21
International Harmonisation
The Organisation for Economic Cooperation and Development (OECD ) formulated the first worldwide OECD Principles of GLP ( revised in 1997) - to avoid non-tariff barriers to trade - to promote mutual acceptance to non-clinical safety test to eliminate unneccessary duplication of experiments International harmonisation of tests Adherence of member countries to these OECD standards permits international acceptability of safety testing from different countries (1981). Malaysia became a provisional member in 1998.
 formulated the first worldwide OECD Principles of GLP 1981 ( revised in 1997) - to avoid non-tariff barriers to trade. - to promote mutual acceptance to non-clinical safety test. to eliminate unneccessary duplication of experiments. International harmonisation of tests. Adherence of member countries to these OECD standards permits international acceptability of safety testing from different countries (1981). Malaysia became a provisional member in")
22
OECD Definition of GLP Good Laboratory Practice (GLP) embodies a set of principles that provides a framework within which laboratory studies are planned, performed, monitored, recorded, reported and archived. These studies are undertaken to generate data by which the hazards and risks to users, consumers and third parties, including the environment, can be assessed for pharmaceuticals (only preclinical studies), agrochemicals, cosmetics, food additives, feed additives and contaminants, novel foods, biocides, detergents etc....
 embodies a set of principles that provides a framework within which laboratory studies are planned, performed, monitored, recorded, reported and archived. These studies are undertaken to generate data by which the hazards and risks to users, consumers and third parties, including the environment, can be assessed for pharmaceuticals (only preclinical studies), agrochemicals, cosmetics, food additives, feed additives and contaminants, novel foods, biocides, detergents etc....")
23
GLP regulations (in US)
Deficiencies made public in the Kennedy Hearings of the US Congress. FDA decide to regulate laboratory testing. GLP is an FDA regulation. Political outcome led to the publication by FDA of Proposed Regulations on Good Laboratory Practice in 1976, Final Rule , June 1979 This forms the regulatory basis for assurance that reports on studies submitted to FDA would reflect faithfully and completely the experimental work carried out.

24
GLP regulations (in Malaysia)
Effective from 29th March 2013, Malaysia is officially a non-member with full adherent to the Organisation for Economic Cooperation and Development (OECD) Council Acts related to Mutual Acceptance of Data (MAD) in the Assessment of Chemicals on Good Laboratory Practice (GLP) GLP compliance is not mandatory but voluntary in Malaysia - certification by NPCB In countries in OECD group such as US, it is required by law that any non-clinical studies must to be conducted in compliance with the OECD Principles of GLP for products to be registered in their countries.
 Council Acts related to Mutual Acceptance of Data (MAD) in the Assessment of Chemicals on Good Laboratory Practice (GLP) GLP compliance is not mandatory but voluntary in Malaysia - certification by NPCB. In countries in OECD group such as US, it is required by law that any non-clinical studies must to be conducted in compliance with the OECD Principles of GLP for products to be registered in their countries.")
25
OBJECTIVES OF GLP GLP makes sure that the data submitted are a true reflection of the results that are obtained during the study. GLP also makes sure that data is traceable. Promotes international acceptance of tests. It does not concern with the technical validity of the studies GLP is sometimes confused with the standards of laboratory safety like wearing safety goggles (Good Laboratory Practices).
.")
27
GLP Regulations: Rules and Tools
Chemical and sample inventory, expiration dates TEST, CONTROL, AND REFERENCE SUBSTANCES Timely reporting, storage of raw data and reports RECORDS AND REPORTS Standard operating procedures FACILITY OPERATION Calibration, logbooks of use, repair, and maintenance EQUIPMENT Maintain adequate space/separation of chemicals from office areas FACILITIES Training records, CVs, GLP training ORGANIZATION AND PERSONNEL Documentation (Tools) GLP Regulations (Rules)
 GLP Regulations (Rules)")
28
Fundamental points of GLP
Good Laboratory Practice applied in whatever industry targeted, stresses the importance of the following main points Resources : Organisation, personnel, facilities, equipment Rules : Protocols, Standard Operating Procedures, concept of Study Director Characterization : Test items, test systems Documentation : Raw data, final report, archives Quality Assurance : Independence from study conduct

29
Resources Organization & Personnel
Structure of org and Org chart must reflect realities and up-to date Responsbilities of all personnel clearly defined; job description, qualifications & competence defined in training and education records. Study Director Full responsibility of GLP compliance of all activities within study, signed compliance statement Aware of all occurrences, judge impact and institute corrective action

30
Facilities and Equipment
Adequate and sufficient to perform the studies Suitable size, construction and location causing minimize disturbances that would interfere with validity of study. Avoid problem of overcrowding, cross contamination, confusion between projects and cramped working condition. Utilities ( water, electricity etc) must be adequate and stable
 must be adequate and stable.")
31
Facilities and Equipment
Separation physically and by organization ensures that different functions or activities do not interfere with one another Important in: Pharmacy and dose mixing areas Histopathology or analytical laboratories – sample mixed up Animal facility- minimize the effects of environmental variables on the animals, facility designed and operated to prevent animals coming in contact with the disease or with a test item other than the one under investigation

32
Facilities and Equipment
Equipment : strict programme of validation, qualification, calibration and maintenance. Records of procedures maintained Reagents: labeled appropriately to indicate source, identity, concentration and stability information, earliest expiratory date (in a kit) and specific storage instructions Separate facilities for handling and storage of test and reference material to prevent contamination and ensure safe storage for hazardous substances
 and specific storage instructions. Separate facilities for handling and storage of test and reference material to prevent contamination and ensure safe storage for hazardous substances.")
33
Facilities and Equipment
Handling and disposal of waste should be carried out in such a manner so as not to influence the integrity of the study in progress and consistent with regulatory requirements Appropriate collection, storage, disposal facilities, decontamination, transportation and destruction procedures Archive facilities should be provided for storage and retrieval of raw data, samples and specimens. Access to archive should be limited to personnel authorized by management

34
Rules Protocols The principle steps of studies have to be described in a Study Protocol or Study Plan The Protocol has to be adopted by the Study Director through dated signature before study starts and alterations to the study design cannot be made unless by formal amendment procedures.

35
Rules Written Procedures
Routine procedures described in Standard Operating Procedures (SOP) Standardization of certain techniques to facilitate comparisons of results Procedures must be reviewed regularly and modified if necessary to reflect the actual state of the art SOPs must be available at the work place and in current version
 Standardization of certain techniques to facilitate comparisons of results. Procedures must be reviewed regularly and modified if necessary to reflect the actual state of the art. SOPs must be available at the work place and in current version.")
36
Rules Study Director Concept of SD as the pivotal point of study control The single most important individual in a GLP study as he/she represents the pivotal point of study control. is the person fully responsible the adequacy of the protocol and the GLP compliant conduct of the study. has to formally accept responsibility for GLP compliance by signing the compliance statement

37
Characterisation Preclinical safety testing of pharmaceutical compunds requires detail knowledge about the properties of the test items and of the test system (often animal ) to which it is administered. Identity, purity, composition, stability, impurity profile should be known for the test item, for the vehicle and for the reference material. If the test system is an animal, it is essential to know such details as its strain, health status and normal biological values
 to which it is administered. Identity, purity, composition, stability, impurity profile should be known for the test item, for the vehicle and for the reference material. If the test system is an animal, it is essential to know such details as its strain, health status and normal biological values.")
38
Documentation Raw Data
Results of investigations, documentation of procedures and circumstances (temperature, pressure, etc) under which study was conducted Results and their interpretation must be a true reflection of the raw data Study Report Responsibility of Study Director Ensures contents of report describe the study accurately Study Director responsible for scientific interpretation of the results
 under which study was conducted. Results and their interpretation must be a true reflection of the raw data. Study Report. Responsibility of Study Director. Ensures contents of report describe the study accurately. Study Director responsible for scientific interpretation of the results.")
39
Raw Data Examples of raw data
If anyone scribble some notes on a scrap of paper, are those notes considered raw data? Examples of raw data Logbooks (to record temperatures or equipment use, repair, and maintenance) Field or laboratory notebooks Forms (for field or laboratory observations, chain-of-custody, sample or chemical receipt) Training reports Computer printouts Recorded data from automated instruments Question: What happens if you make a mistake? Do not obscure original data!! Instead, draw a single strikeout, then add reason code, initials, and date of change.
 Field or laboratory notebooks. Forms (for field or laboratory observations, chain-of-custody, sample or chemical receipt) Training reports. Computer printouts. Recorded data from automated instruments. Question: What happens if you make a mistake Do not obscure original data!! Instead, draw a single strikeout, then add reason code, initials, and date of change.")
40
Documentation Archives / Retention of records & materials
For reasons of reconstruction & traceability many years later (usually 7 years; medical records: 20years) Safekeeping of all records, kept in integral state and can neither be lost nor altered Restrict access to archives to a limited number of people and maintain records of log-in and log-out for both documents and people
 Safekeeping of all records, kept in integral state and can neither be lost nor altered. Restrict access to archives to a limited number of people and maintain records of log-in and log-out for both documents and people.")
41
Quality Assurance The test facility should have a documented Quality Assurance Programme to assure that studies performed are in compliance with these Principles of GLP. The Quality Assurance Programme should be carried out by an individual or by individuals designated by and directly responsible to management and who are familiar with the test procedures. QA personnel has to be independent of the operational conduct of the studies and it functions as witness to the whole preclinical research process

42
ISO Accreditation vs GLP / GMP compliance
Fundamental requirement of the GLP Principles not covered in ISO/IEC : the use of study plans and Study Director as a concept More stringent under GLP - Recording and reporting of data - Management of data retained in archive to allow complete reconstruction of study - A program of independent QA including internal audits of every study

43
ISO Accrediting Bodies for Malaysia
ISO compliance: technical assessment by Accrediting Body Standards Malaysia – ISO 15189, 17025 SIRIM QAS Internation – ISO 9001 Registration Authorities looks for GMP & GLP compliances Drug Control Authority or National Pharmaceutical Control Bureau

44
OECD GLP Compliance Monitoring Authorities for Malaysia
Pharmaceutical products Cosmetics Food additive products Veterinary Pesticides Industrial products Feed Additive products Biotechnology (non- pharmaceutical) products National Pharmaceutical Control Bureau (NPCB) MoH STANDARDS MALAYSIA, MOSTI
 products. National Pharmaceutical Control Bureau (NPCB) MoH. STANDARDS MALAYSIA, MOSTI.")
45
Thank You Say what you do, do what you say, prove it and improve it
Janet Woodcook, M.D. Director, Center for Drug Evaluation and Research, FDA Record what you do, not recorded not done "If it's not written down, it didn't happen” Thank You

Similar presentations










>")









