Download presentation
Presentation is loading. Please wait.

1
Ab initio REMPI Erlendur Jónsson

2
MSc project Electronically excited states of HX(H 2 O) n After some trial calculations, this morphed into just calculations of HF and later on HCl
 n After some trial calculations, this morphed into just calculations of HF and later on HCl")
3
Calculations The calculations I’ve been using are all approximate methods of solving the Schrödinger equation

4
Calculations The excited-state calculations are apparently very hard. The methods that are used for them are –TD-DFT –CI –CC

5
TD-DFT Time-dependent density functional theory The cheapest method Results are highly dependent on the selection of functional Doesn’t handle non-Rydberg character properly

6
CI Configuration interaction Handles correlated electrons Can be formally exact Extremely expensive Common approximation is the CISD –Configuration interaction singles doubles

7
CC Coupled cluster Can be formally exact like CI, but cheaper CCSD(T) is currently the gold-standard of quantum chemistry
 is currently the gold-standard of quantum chemistry")
8
CC The S is singly excited electron The D are double excited electrons A parenthesis, like (T), means that triple excitations are partially calculated via pertubation Implementations exist for up to CCSDTQPH
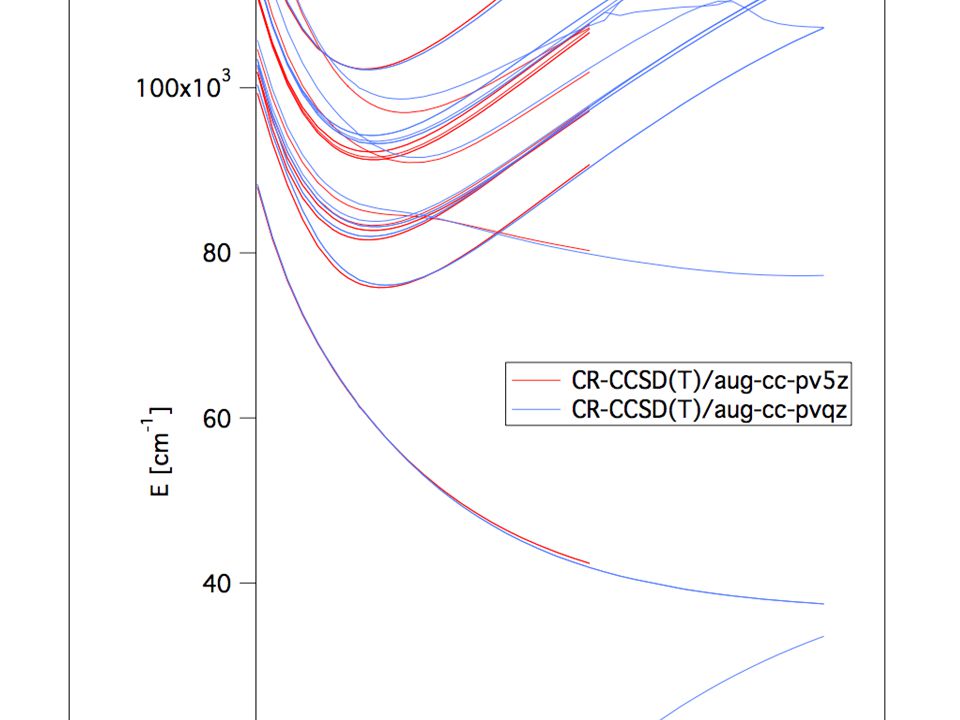
, means that triple excitations are partially calculated via pertubation Implementations exist for up to CCSDTQPH")
9
CC - excited states EOMCC –Equations of motions coupled cluster Fairly reliable A lot of research being done at the moment in new methods and extensions of the old methods

10
CC CC methods have a hard time handling bond breaking and high inter-nuclear distance To compensate, new extensions have been added, such as the LR-CC and CR-CC (locally and completely renormalized)
")
11
Bases Systematic basis sets such as the cc-pvNz basis of Dunning, et al. give a very convenient way to improve calculations But to handle very electronegative atoms, such as fluorine and chlorine, diffuse functions are needed in the basis which aren’t in the cc-pvNz so I’ve used the aug-cc-pvNz

12
aug-cc-pvNz Augmented correlation consistent polarized valence N zeta N can be Double, Triple, Quadruple, 5 (quintuple) or 6 (sextuple) Very popular for estimation of Complete Basis Set limit
 or 6 (sextuple) Very popular for estimation of Complete Basis Set limit")
13
aug-cc-pcvNz Extension of the aug-cc-pvNz where more core-core and core-valence correlation effects are added When I tried excited triplet state calculations they proved to work considerably better than the aug-cc- pvNz

14
HF Was able to get fairly good results The usual EOMCCSD calculations weren’t able to handle the V state of HF Needed CR-EOM-CCSD(T) But when that was achieved, the experimental setup didn’t work properly so I started calculations for HCl
 But when that was achieved, the experimental setup didn’t work properly so I started calculations for HCl")
15
Method X 1 Σ + r e [Å]ω e [cm -1 ] B 1 Σ + r e [Å]ω e [cm -1 ] CCSD/aug-cc-pVDZ CCSD/aug-cc-pVTZ CCSD/aug-cc-pVQZ CCSD/d-aug-cc-pVDZ CCSD/d-aug-cc-pVTZ CCSD/d-aug-cc-pVQZ 0.92193 0.91642 0.91384 0.92151 0.91669 0.91380 4116.9 4184.7 4211.3 4111.3 4186.0 4201.2 1.98837 1.98046 1.98791 1.98987 1.97995 1.98666 1127.7 1101.9 1098.9 1127.0 1102.1 1099.4 CCSD(T)/aug-cc-pv5Z[1] MRD-CI[3] Exp[2] 0.9173 0.92311 0.91680 4141.9 4148.64 4138.32 --- 2.1516 2.0908 --- 1131.2 1159.18 [1]K.A. Peterson and T.H. Dunning, J. Chem. Phys. 102, 2032,1995 [2] Retrieved from http://webbook.nist.govhttp://webbook.nist.gov [3] Bettendorff, M.,et al. Zeitschrift Fur Physik a-Hadrons and Nuclei, 304, 125-135, 1982
![Method X 1 Σ + r e [Å]ω e [cm -1 ] B 1 Σ + r e [Å]ω e [cm -1 ] CCSD/aug-cc-pVDZ CCSD/aug-cc-pVTZ CCSD/aug-cc-pVQZ CCSD/d-aug-cc-pVDZ CCSD/d-aug-cc-pVTZ CCSD/d-aug-cc-pVQZ CCSD(T)/aug-cc-pv5Z[1] MRD-CI[3] Exp[2] [1]K.A.](http://images.slideplayer.com/16/5119537/slides/slide_15.jpg "Peterson and T.H. Dunning, J. Chem. Phys. 102, 2032,1995 [2] Retrieved from [3] Bettendorff, M.,et al. Zeitschrift Fur Physik a-Hadrons and Nuclei, 304, ,")
17
HCl Harder than HF –More electrons –Larger basis I’ve used the experience gained from HF to progress further into the HCl calculations

18
HCl Is C ∞v group, but the programs only offer C 2v This means that the excited state symmetries are a1, a2, b1 and b2 Which aren’t the real symmetries which we have been seeking So it hasn’t been easy finding what state is what in the resulting calculations

19
HCl Our hypothesis is that a1 states have Σ symmetry, a2 Δ symmetry and b1 have Π symmetry b1 and b2 are degenerate

20
Experimental vs. calculations We of course need to compare the ab initio calculations to experimental results The problems is that we have a potential curve

22
Experimental vs. calculations Currently we just fit the potential and get the various spectroscopic parameters These parameters can then be used to simulate a REMPI spectra

23
T0T0 DeDe rere ee exeexe F1F1 CR/5Z82659347891.3082786.1455.78 CR/QZ83178469841.3332617.7336.46 SD/QZ81270335261.3212715.1954.97 Experimental82847.3 [1]34464.18 [3]1.295 [3]2608.3 [3]49.35 [3] C1C1 CR/5Z75868408221.3352847.4149.65 CR/QZ76174440001.3512766.1143.47 SD/QZ76266560021.3492840.2536.01 Experimental77485.2 [1]---1.358 [2]2684.0 [2]66.0 [2] X1+X1+ CR/5Z-1511.1441031.2703090.3154.13 CR/QZ-1533.5452881.2773043.0951.12 SD/QZ-1559.2453381.2773047.5951.21 Experimental-1482.2685 [1] 42330 [1]1.27455 [2]2990.946 [2] 52.8186 [2]
![T0T0 DeDe rere ee exeexe F1F1 CR/5Z CR/QZ SD/QZ Experimental [1] [3]1.295 [3] [3]49.35 [3] C1C1 CR/5Z CR/QZ SD/QZ Experimental [1] [2] [2]66.0 [2] X1+X1+ CR/5Z CR/QZ SD/QZ Experimental [1] [1] [2] [2] [2]](http://images.slideplayer.com/16/5119537/slides/slide_23.jpg "T0T0 DeDe rere ee exeexe F1F1 CR/5Z CR/QZ SD/QZ Experimental [1] [3]1.295 [3] [3]49.35 [3] C1C1 CR/5Z CR/QZ SD/QZ Experimental [1] [2] [2]66.0 [2] X1+X1+ CR/5Z CR/QZ SD/QZ Experimental [1] [1] [2] [2] [2]")
25
The future Automate the simulation of the REMPI spectra and if possible remove the fitting part of method –Make a ab initio REMPI simulator

26
Thank you for your attention

Similar presentations



 Workshop.>")














 assumptions are made, and that the method.>")


