Download presentation
Presentation is loading. Please wait.

1
Myeloproliferative disorders
Clonal hematopoiesis (stem cell disorder) Marrow hypercellularity Overproduction of one or more cell lines (effective hematopoiesis) Exception: myelofibrosis Differentiation nearly normal
 Marrow hypercellularity. Overproduction of one or more cell lines (effective hematopoiesis) Exception: myelofibrosis. Differentiation nearly normal.")
2
The “common” bcr-abl negative myeloproliferative disorders
Polycythemia vera Essential thrombocythemia Myelofibrosis/myeloid metaplasia

3
Less common conditions
Undifferentiated myeloproliferative disorder Mast cell disease Hypereosinophilic syndrome Chronic neutrophilic leukemia Myelodysplastic/myeloproliferative disorders CMML Atypical CML (BCR-ABL negative) Juvenile CML “Unclassifiable” MDS/MPD
 Juvenile CML. Unclassifiable MDS/MPD.")
4
JAK-2 Tyrosine kinase involved in signaling pathways initiated by EPO, TPO, G-CSF and other growth factors V617F mutations in most cases of PV, ET, MF Other JAK-2 mutations (exon 12, etc) in a minority of PV cases (almost all cases of PV have a JAK-2 mutation of some type) Some cases of ET, MF lack JAK-2 mutation Mutation releases cells from dependence on growth factors Homozygosity for mutation seen mainly in P vera, associated with disease progression Specific inhibitors of the kinase now in clinical trials
 in a minority of PV cases (almost all cases of PV have a JAK-2 mutation of some type) Some cases of ET, MF lack JAK-2 mutation. Mutation releases cells from dependence on growth factors. Homozygosity for mutation seen mainly in P vera, associated with disease progression. Specific inhibitors of the kinase now in clinical trials.")
5
Structure-function relationships in the JAK2 receptor
Science 2014; 344:703

6
In Panel A, in the absence of ligand, the erythropoietin receptor (EPOR) binds JAK2 as an inactive dimer. In cells with wild-type JAK2 protein, the binding of erythropoietin (Epo) to its receptor induces conformational changes in the receptor, resulting in phosphorylation (P) of JAK2 and the cytoplasmic tail of the receptor. This leads to signaling through pathways made up of Janus kinases and signal transducers and activators of transcription (JAK–STAT), phosphatidylinositol 3 kinase (PI3K), and RAS and mitogen-activated protein kinase (RAS–MAPK). In cells with the V617F mutation, the signaling is constitutively increased, even in the absence of erythropoietin. In Panel B, the JAK2 protein binds to multiple cytokine receptors — EPOR, thrombopoietin receptor (MPL), granulocyte colony-stimulating factor receptor (G-CSFR), and probably others — that are important for hematopoietic stem-cell biology and differentiation. Therefore, the JAK2 protein with the V617F mutation exerts its effects at various stages of differentiation and in various lineages. In Panel C, the development of homozygosity for the V617F mutation is a two-step process, with the initial point mutation followed by mitotic recombination of chromosome 9p between the JAK2 locus and the centromere. This results in the loss of heterozygosity but a diploid DNA copy number. NEJM 2006; 355:2452

8
NEJM 2006; 355:2452

9
Polycythemia vera Elevated RBC mass, typically high platelets and WBC
Some patients present with thrombocytosis and develop erythrocytosis subsequently Hypercellular marrow with variable degree of reticulin fibrosis Morphology fairly normal, some clustering and dysmorphism of megas Splenomegaly in 70%, constitutional sx, increased risk of arterial & venous thrombosis (esp portal vein), microvascular disease (erythromelalgia, pruritus, headache, etc), hypermetabolic sx & gout
, microvascular disease (erythromelalgia, pruritus, headache, etc), hypermetabolic sx & gout.")
10
Differential diagnosis of erythrocytosis
H&P COPD or other possible causes of hypoxemia? Smoker? Splenomegaly, pruritus, erythromelalgia? Concomitant thrombocytosis and/or leukocytosis? Serum EPO JAK-2 mutation testing

11
Polycythemia Vera Natural History
Life expectancy decreased (3 deaths/100 pts/yr) Cardiovascular mortality increased 1.4x Major thrombosis in 3/100pts/yr Risk of death from leukemia increased 36-fold Progressive myelofibrosis
 Cardiovascular mortality increased 1.4x. Major thrombosis in 3/100pts/yr. Risk of death from leukemia increased 36-fold. Progressive myelofibrosis.")
12
NEJM 2004; 350:99

13
NEJM 2004; 350:99

14
Oxygen delivery vs Hematocrit
J Clin Invest 1963;42:1150

15
Vaso-occlusion in P vera

16
Thrombosis in myeloproliferative disorders
41% of deaths in PV from cardiovascular causes 15% coronary dz 8% CHF 8% non hemorrhagic stroke 8% PE Complex pathophysiology Abnormal RBC/WBC/plt function Activated PMNs/cytokines affect vascular endothelium Prothrombotic microparticle release Increased whole blood viscosity

17
Risk factors for thrombosis in myeloproliferative disorders
Hx of thrombosis Disease category (PV > ET, MF) Cardiovascular risk factors (lipids, blood pressure, smoking, diabetes) Age OCP therapy (splanchnic vein clots) JAK2 mutation status/allele burden High hematocrit (PV) & WBC Extreme thrombocytosis increases bleeding risk Thrombophilic genetic traits?
 Cardiovascular risk factors (lipids, blood pressure, smoking, diabetes) Age. OCP therapy (splanchnic vein clots) JAK2 mutation status/allele burden. High hematocrit (PV) & WBC. Extreme thrombocytosis increases bleeding risk. Thrombophilic genetic traits")
18
Prothrombotic effects of erythrocytosis
Barbui et al. Blood 2013;122:

19
Hematology 2005:201

20
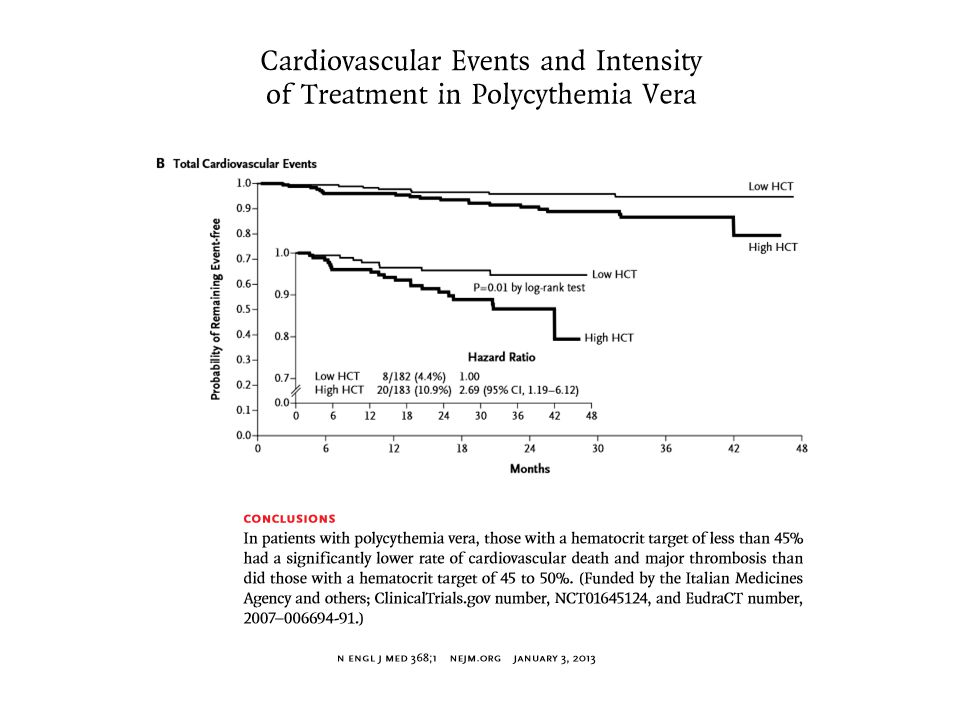
Recommendations for treatment of patients with polycythemia vera
Keep Hct < 45 (plebotomy) Low dose ASA unless contraindicated Manage reversible risk factors (BP, lipids, smoking) Consider cytoreduction if: Intolerant to phlebotomy Thrombocytosis or substantial leukocytosis Symptomatic splenomegaly Choice of cytoreductive therapy: Age < 40: Interferon-alpha Age > 40: hydroxyurea Anagrelide or busulfan for patients intolerant of, or not responsive to, above choices Hematology 2005:201
 Low dose ASA unless contraindicated. Manage reversible risk factors (BP, lipids, smoking) Consider cytoreduction if: Intolerant to phlebotomy. Thrombocytosis or substantial leukocytosis. Symptomatic splenomegaly. Choice of cytoreductive therapy: Age < 40: Interferon-alpha. Age > 40: hydroxyurea. Anagrelide or busulfan for patients intolerant of, or not responsive to, above choices. Hematology 2005:201.")
22
Aspirin vs placebo in P vera

23
Essential thrombocytosis
Thrombocytosis (typically > 600K) with variable leukocytosis, normal RBC count About 50% have JAK2 mutation Normal to mildly hypercellular marrow with marked increase in megakaryocytes, often with clustering Minimal reticulin fibrosis Splenomegaly in about 50% Many patients asymptomatic at diagnosis Increased risk of arterial and venous thrombosis as well as hemorrhage
 with variable leukocytosis, normal RBC count. About 50% have JAK2 mutation. Normal to mildly hypercellular marrow with marked increase in megakaryocytes, often with clustering. Minimal reticulin fibrosis. Splenomegaly in about 50% Many patients asymptomatic at diagnosis. Increased risk of arterial and venous thrombosis as well as hemorrhage.")
24
Thrombocytosis Differential diagnosis
Essential thrombocythemia Other MPD P vera Myelofibrosis CML Myelodysplasia (5q- et al) Reactive Inflammation Surgery/trauma Non-myeloid malignancy Iron deficiency Hemolysis Acute blood loss Absence of spleen Rebound following thrombocytopenia
 Reactive. Inflammation. Surgery/trauma. Non-myeloid malignancy. Iron deficiency. Hemolysis. Acute blood loss. Absence of spleen. Rebound following thrombocytopenia.")
25
Hematology 2005:201

26
Essential Thrombocytosis Natural History
1-2 cases/100,000/yr Most patients >50, but occurs in young adults Woman more often affected than men Life expectancy normal to slightly decreased Patients > 60, or with prior thrombosis, at increased risk for thrombotic events Very high platelet counts (>1.5 million) increase risk of bleeding, not thrombosis Risk of myelofibrosis about 8% at 10 years Risk of AML about 1% overall
 increase risk of bleeding, not thrombosis. Risk of myelofibrosis about 8% at 10 years. Risk of AML about 1% overall.")
27
Essential Thrombocytosis Treatment
Low-risk patients (young, no vascular risk factors) may not require treatment Aspirin decreases thrombotic events Hydroxyurea Anagrelide Interferon Other: busulfan (leukemogenic)
 may not require treatment. Aspirin decreases thrombotic events. Hydroxyurea. Anagrelide. Interferon. Other: busulfan (leukemogenic)")
28
Recommendations for treatment of patients with essential thrombocytosis
Manage reversible risk factors (BP, lipids, smoking) If hx of thrombosis, or age >60, or plts > 1.5 million: Hydroxyurea Low dose ASA unless for plts > 1.5 m 2nd line: Anagrelide or interferon-alpha Age < 60, no other risk factors: Low dose ASA Hematology 2005:201
 If hx of thrombosis, or age >60, or plts > 1.5 million: Hydroxyurea. Low dose ASA unless for plts > 1.5 m. 2nd line: Anagrelide or interferon-alpha. Age < 60, no other risk factors: Low dose ASA. Hematology 2005:201.")
30
Myelofibrosis 0.5-1.5 cases/100,000/yr Most patients >60
Marrow fibrosis with extramedullary hematopoiesis in spleen, liver, many other tissues “Prefibrotic” stage with hypercellular marrow & abnormal megakaryocytes Peripheral leukoerythroblastosis, teardrop cells, often with anemia. WBC and platelet counts may be high, normal or low Splenomegaly, constitutional symptoms Poor prognosis: marrow failure, AML in 5-30%

31
Improving survival in MF
Median survival 6.5 yrs Median survival 4.5 yrs J Clin Oncol 2012;30:2981

32
DDx of Marrow Fibrosis Myeloproliferative disorders
MF > > P vera, ET, CML Other heme neoplasm Megakaryocytic leukemia Hodgkins Hairy cell leukemia Non-heme cancer Non-malignant conditions Renal osteodystrophy Autoimmune disease Vitamin D deficiency

33
SPLENOMEGALY IN MYELOFIBROSIS
Mayo Clin Proc 2004;79:503 SPLENOMEGALY IN MYELOFIBROSIS

34
Myelofibrosis Treatment
Supportive care Transfusions Splenectomy Thalidomide/lenalidomide (low dose) Reduce transfusion requirements, reduce spleen size Chemotherapy (limited data; myelosuppression often dose-limiting) Cladribine Azacytidine/decitabine Low dose cytarabine Marrow transplant in selected patients JAK inhibitor therapy: Ruxolitinib (inhibits JAK-1 and JAK-2)
 Reduce transfusion requirements, reduce spleen size. Chemotherapy (limited data; myelosuppression often dose-limiting) Cladribine. Azacytidine/decitabine. Low dose cytarabine. Marrow transplant in selected patients. JAK inhibitor therapy: Ruxolitinib (inhibits JAK-1 and JAK-2)")
35
Ruxolitinib treatment of myelofibrosis
Spleens shrink Symptoms improve Better survival NEJM 2012;366:799

36
Eosinophilia Non-clonal/secondary (common) Clonal
Idiopathic hypereosinophilic syndrome Familial (very rare) Mild: AEC Moderate: AEC Severe: AEC > 5000
 Mild: AEC Moderate: AEC Severe: AEC >")
37
Hypereosinophilia (AEC > 5000)
Parasitic infection Drug reaction Allergic gastroenteritis Eosinophilic fasciitis Churg-Strauss syndrome Pulmonary eosinophilia Eosinophilia-myalgia syndrome IL-2 therapy Hyper-IgE syndrome AML Eosinophilic leukemia Hypereosinophilic syndrome

38
Clinical manifestations of hypereosinophilia
Skin: pruritus, angioedema, ulcers, papular or nodular lesions Heart: endocardial fibrosis, valvular disease, mural thrombi, cardiomyopathy, incr troponin Lungs: infiltrates, nodules, pleural effusion Neurologic: poly- or mononeuropathy, transverse myelitis, optic neuritis, CNS vasculitis GI: hepatosplenomegaly, gastroenteritis, sclerosing cholangitis Heme: marrow fibrosis, cytopenias Kidney: thrombotic microangiopathy

39
Secondary Eosinophilia
Infection Usually parasitic Allergy Asthma, allergic rhinitis, dermatitis, urticaria/angioedema, drug reaction Autoimmune/inflammatory disorders (many) Paraneoplastic Solid tumors, lymphomas (Hodgkin>NHL) Endocrinopathy Adrenal insufficiency, growth hormone deficiency Clonal T-cell disorder (→ IL-5) Most cases with IL-5 overproduction
 Paraneoplastic. Solid tumors, lymphomas (Hodgkin>NHL) Endocrinopathy. Adrenal insufficiency, growth hormone deficiency. Clonal T-cell disorder (→ IL-5) Most cases with IL-5 overproduction.")
40
Clonal Eosinophilia Acute leukemia (AML>ALL)
Chronic myeloid disorders CML (bcr-abl) Systemic mastocytosis (c-Kit) 8p11 myeloproliferative disorder (FGFR-1) PDGFRA and PDGFRB mutations “Classic” MPD (JAK-2) MDS Chronic eosinophilic leukemia
 Systemic mastocytosis (c-Kit) 8p11 myeloproliferative disorder (FGFR-1) PDGFRA and PDGFRB mutations. Classic MPD (JAK-2) MDS. Chronic eosinophilic leukemia.")
41
Clonal eosinophilic disorders caused by mutations in tyrosine kinase genes
PDGFRA, PDGFRB May be cytogenetically silent→ FISH testing for dx Marrow fibrosis, incr mast cells, elevated tryptase → variant of mast cell dz? Male predominance Imatinib-responsive ( mg/d) FGFR1 Associated with 8p11 translocations Stem cell disorder → aggressive MPD associated with T-lymphoblastic lymphoma High dose chemotherapy + allo-HSCT
 FGFR1. Associated with 8p11 translocations. Stem cell disorder → aggressive MPD associated with T-lymphoblastic lymphoma. High dose chemotherapy + allo-HSCT.")
42
Chronic eosinophilic leukemia WHO diagnostic criteria
Eosinophil count > 1500 No Ph chromosome or BCR-ABL fusion No PDGFRB, PDGFRA or FGFR1 rearrangement Blast count in blood and marrow < 20% No inv (16) or other variant characteristic of AML Presence of clonal cytogenetic or molecular abnormality OR blasts >2% in blood or >5% in marrow
 or other variant characteristic of AML. Presence of clonal cytogenetic or molecular abnormality OR blasts >2% in blood or >5% in marrow.")
43
Idiopathic Hypereosinophilic Syndrome
Persistent moderate or severe eosinophilia (AEC > 1500) with end-organ damage Other causes of eosinophilia ruled out Male predominance Can evolve into overt myeloid or lymphoid neoplasm Potentially fatal – 10 yr survival < 50% Treatment Asymptomatic: corticosteroids vs watchful waiting Symptomatic patients: corticosteroids, IFNα, hydroxyurea, imatinib, various myelosuppressive agents
 with end-organ damage. Other causes of eosinophilia ruled out. Male predominance. Can evolve into overt myeloid or lymphoid neoplasm. Potentially fatal – 10 yr survival < 50% Treatment. Asymptomatic: corticosteroids vs watchful waiting. Symptomatic patients: corticosteroids, IFNα, hydroxyurea, imatinib, various myelosuppressive agents.")
44
Laboratory evaluation of hypereosinophilia
Marrow biopsy with cytogenetics FISH for PDGFR mutations Serum tryptase T-cell immunophenotyping and gene rearrangement study Serum IL-5 Serum IgE Screen for organ damage Echo, troponin level CXR, PFTs

45
Classification of Mast Cell Disease WHO classification and frequency in Mayo Clinic series
Indolent – often limited to skin (urticaria pigmentosa (46%) Associated with myeloproliferative disorder (40%) “Aggressive” mastocytosis – with lymphadenopathy & eosinophilia (12%) Mast cell leukemia (1%) Blood 2009;113:5727
 Associated with myeloproliferative disorder (40%) Aggressive mastocytosis – with lymphadenopathy & eosinophilia (12%) Mast cell leukemia (1%) Blood 2009;113:5727.")
46
WHO Diagnostic Criteria for Mast Cell Disease
Major: multifocal dense infiltrates of mast cells (15+) in marrow or other extracutaneous organs Minor >25% spindle shaped or otherwise atypical MC c-KIT codon 816 mutation MC in marrow or other extracutaneous organ express CD2 and/or CD25 Serum tryptase persistently >20 ng/ml in the absence of another clonal myeloid disorder Diagnosis requires major + 1 minor criterion or at least 3 minor criteria
 in marrow or other extracutaneous organs. Minor. >25% spindle shaped or otherwise atypical MC. c-KIT codon 816 mutation. MC in marrow or other extracutaneous organ express CD2 and/or CD25. Serum tryptase persistently >20 ng/ml in the absence of another clonal myeloid disorder. Diagnosis requires major + 1 minor criterion or at least 3 minor criteria.")
47
Stuff in mast cell granules that could make you sick
Metcalfe, Blood 2008;112:946

48
Clinical & laboratory features of mast cell disease
Skin: Urticaria pigmentosa, Darier sign (dermatographism), telangiectasias Lymphadenopathy Hepatosplenomegaly Constitutional symptoms Recurrent anaphylaxis GI symptoms (N/V, diarrhea, abd pain, peptic ulcer) Elevated serum/urinary histamine levels Elevated serum tryptase (96%) Cytopenias Eosinophilia Coagulopathy (heparin in mast cells) – rare Lytic or blastic bone lesions
, telangiectasias. Lymphadenopathy. Hepatosplenomegaly. Constitutional symptoms. Recurrent anaphylaxis. GI symptoms (N/V, diarrhea, abd pain, peptic ulcer) Elevated serum/urinary histamine levels. Elevated serum tryptase (96%) Cytopenias. Eosinophilia. Coagulopathy (heparin in mast cells) – rare. Lytic or blastic bone lesions.")
49
Blood 2009; 113: 5727 ISM: Indolent MCD SM-ANMD: associated with clonal non-mast cell lineage disease ASM: Aggressive MCD MCL: Mast cell leukemia

50
Mastocytosis in bone marrow
H&E Tryptase

51
Mastocytosis in bone marrow

52
Skin findings in mast cell disease
Dermatographism Urticaria pigmentosa Telangiectasia macularis eruptiva perstans

53
Molecular biology of mast cell disease
A majority of patients have D816V mutation in c-KIT tyrosine kinase gene (imatinib-insensitive) A minority have other mutations, may be imatinib-sensitive Occasionally found together with JAK2 V617F Some patients have eosinophilia & PDGFRA mutation (imatinib-responsive)
 A minority have other mutations, may be imatinib-sensitive. Occasionally found together with JAK2 V617F. Some patients have eosinophilia & PDGFRA mutation (imatinib-responsive)")
54
Treatment of mast cell disease
Antihistamines, PPIs Vit D/Calcium, bisphosphonates Glucocorticoids Epinephrine (for anaphyactic reactions) For aggressive disease: Interferon Imatinib (if there is an imatinib-sensitive mutation) Other TKIs (dasatinib, nilotinib)? Cladribine Combination chemotheapy Allo-HSCT
 For aggressive disease: Interferon. Imatinib (if there is an imatinib-sensitive mutation) Other TKIs (dasatinib, nilotinib) Cladribine. Combination chemotheapy. Allo-HSCT.")
55
Survival of patients with MCD
Blood 2009; 113: 5727

Similar presentations







II Dr. Ibrahim. A. Adam.>")

>")

 Myelodysplastic / myeloproliferative diseases (MDS/MPD) >")
:>")







 - Essential Thrombocythemia - Myelofibrose Myeloid Methaplasia.>")
