Download presentation
Presentation is loading. Please wait.

1
[Gly-X-Y]n ‘R’ US READ Gelehrter et al. pages 143-150
SEE course web pages for related objectives and lecture notes
![[Gly-X-Y]n ‘R’ US READ Gelehrter et al. pages](http://slideplayer.com/slide/4556286/15/images/1/%5BGly-X-Y%5Dn+%E2%80%98R%E2%80%99+US+READ+Gelehrter+et+al.+pages.jpg "SEE course web pages for related objectives and lecture notes.")
2
Collagen Disorders Acquired/Multifactorial Aging Mendelian Inheritance
Osteogenesis imperfecta (Type I Collagen) Ehlers-Danlos syndromes (Types I, II,III, V Collagens) Chondrodysplasias (Type II Collagen) Alport syndrome (Type IV Collagen) Stickler syndrome (Types II, XI Collagen) Schmid metaphyseal dysplasia (Type X Collagen) Dystrophic epidermolysis bullosa (Type VII Collagen)
 Ehlers-Danlos syndromes (Types I, II,III, V Collagens) Chondrodysplasias (Type II Collagen) Alport syndrome (Type IV Collagen) Stickler syndrome (Types II, XI Collagen) Schmid metaphyseal dysplasia (Type X Collagen) Dystrophic epidermolysis bullosa (Type VII Collagen)")
3
Arterial rupture 32 68 60 55 53 30s 1 4 5 36 33 31 5 Aortic dissection
HTN Splenic Artery rupture, Vertebral artery rupture 57, Aortic dissection 60, 1 4 5 36 33 31 R internal carotid dissection, cerebral infarct, R vertebral artery aneurysm, s/p repair Abdominal aorta aneurysm/chronic dissection, Aneurysm R common iliac 5 Arterial rupture

4
Procollagen Proelastin Connective tissue proteins are synthesized in a tissue specific manner by a variety of specialized cells such as fibroblasts, chondrocytes, osteoblasts.

5
What is collagen? Major extracellular fibrous protein that provides strength and structure to tissues by organizing the extracellular matrix via supramolecular assembly functions A ropelike structure made by intertwining three polypeptide chains into a triple helix

6
Fibrillar collagens have awesome tensile strength
Reticular collagens provide a meshwork of support

7
25 distinct collagen molecules vary by content of genetically distinct a-chains
Type I collagen: [a1(I)]2a2(I) Heterotrimer two a-1 chains (COLIA1 on chromosome 17) one a-2 chain (COLIA2 on chromosome 7) Type III collagen: [a1(III)]3 Homotrimer two a-1 chains (COL3A1 on chromosome 2)
]2a2(I) Heterotrimer. two a-1 chains (COLIA1 on chromosome 17) one a-2 chain (COLIA2 on chromosome 7) Type III collagen: [a1(III)]3. Homotrimer. two a-1 chains (COL3A1 on chromosome 2)")
8
Skin contains Type I, III, V collagens and elastic fibers
Connective tissue supporting hollow organs including colon contains Type III collagen Connective tissue surrounding blood vessels includes type III collagen and elastic fibers

9
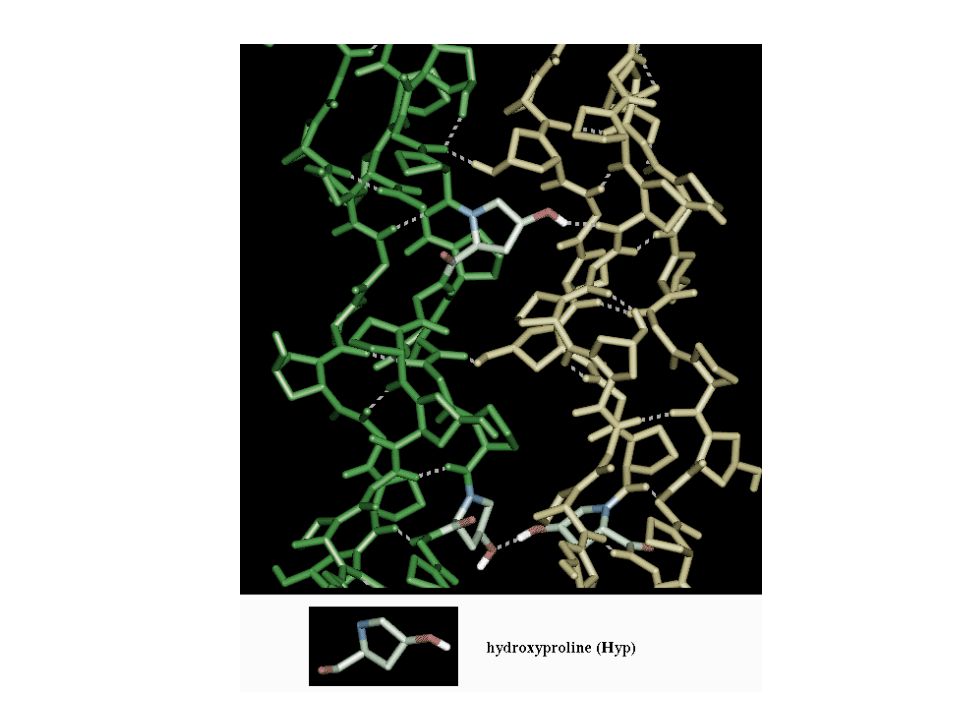
[Gly-X-Y]n Each polypeptide chain is wound in a left-handed helix
X: proline, lysine Y: hydroxyproline, hydroxylysine. Each polypeptide chain is wound in a left-handed helix Three chains form a triple helix monomer The helix is stabilized by hydrogen bonds in a right-handed helix. Importantly, the third residue is glycine, as it can fit into the center of the helix.
![[Gly-X-Y]n Each polypeptide chain is wound in a left-handed helix](http://slideplayer.com/slide/4556286/15/images/9/%5BGly-X-Y%5Dn+Each+polypeptide+chain+is+wound+in+a+left-handed+helix.jpg "X: proline, lysine. Y: hydroxyproline, hydroxylysine. Each polypeptide chain is wound in a left-handed helix. Three chains form a triple helix monomer. The helix is stabilized by hydrogen bonds in a right-handed helix. Importantly, the third residue is glycine, as it can fit into the center of the helix.")
11
disulfide bonds

15
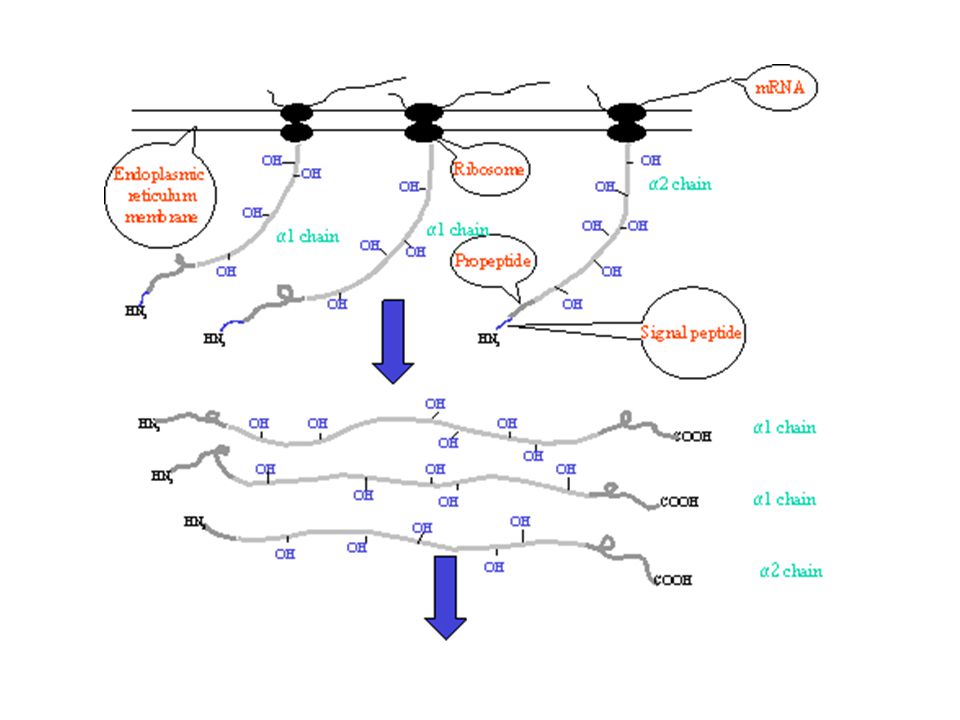
The making of collagen…
Polypeptide chains are synthesized from ribosomes, signal peptides are removed and a series of post-translational modifications take place: Intracellular modifications: Hydroxylation of proline and lysines allows bonding to other molecules which requires co-factors (i.e. vitamin C). Glycosylation of hydroxylysine residues via galactosyl transferase, lysyl hydroxylase, and glucosyl transferase controls fibrillar morphology. The three chains align and disulfide bonds form to form triple helix. Triple helix is packaged into a vesicle and secreted from the cell. Extracellular modifications: Calcium dependent N- and C-proteinases remove the propeptides Procollagen molecules aggregate by a 1/4 staggering in respect to its neighbor and are stabilized by covalent cross links. A polymeric fibril is formed. Fibril is arranged in different tissue specific ways
. Glycosylation of hydroxylysine residues via galactosyl transferase, lysyl hydroxylase, and glucosyl transferase controls fibrillar morphology. The three chains align and disulfide bonds form to form triple helix. Triple helix is packaged into a vesicle and secreted from the cell. Extracellular modifications: Calcium dependent N- and C-proteinases remove the propeptides. Procollagen molecules aggregate by a 1/4 staggering in respect to its neighbor and are stabilized by covalent cross links. A polymeric fibril is formed. Fibril is arranged in different tissue specific ways.")
16
By EM (50,000 X), collagen appears as regular striated bundles in longitudinal section and circles on cross section Type I collagen fibers

17
Some collagens that form long structural fibrils
I. Skin, tendon, bone, dentin; a fetal form also exists Mutations cause osteogenesis imperfecta, procollagen cleavage mutations cause EDS II. Cartilage, vitreous humor Mutations cause abnormalities of skeleton and eye, Stickler syndrome III. With type I in skin, also in muscles, blood vessels Mutations cause Vascular EDS V. Fetal tissues, placenta, interstitial tissues, skin Important for extracellular matrix assembly, mutations cause classic C Classic EDS

18
Type I collagen 90% of our collagen is type I two a1(I) chains (COL1A1 chr 17) and one a2(I) chains (COL1A2 chr 7)) Most abundant in: Bones, skin, tendons, cornea 300nm long 1.5nm in diameter a1(I) a2(I) COL1A1 and COL1A2 mutations cause Osteogenesis Imperfecta syndromes Posttranslational modification defects cause rare Ehlers-Danlos syndromes
 a2(I) COL1A1 and COL1A2 mutations cause Osteogenesis Imperfecta syndromes. Posttranslational modification defects cause rare Ehlers-Danlos syndromes.")
19
Type I collagen fibers arranged to provide structural integrity

20
Osteogenesis Imperfecta
Four major clinical sub-types (Sillence classification) vary in phenotypic characteristics and severity (I,IV,III,II) Highly penetrant, inter- and intra-familial variable expressivity Prognosis varies greatly depending on type
 vary in phenotypic characteristics and severity (I,IV,III,II) Highly penetrant, inter- and intra-familial variable expressivity. Prognosis varies greatly depending on type.")
21
OI Type I (with or without DI)
Most common form Type I collagen is decreased in amount, generally normal in structure Normal/near normal height Little or no bone deformity Blue/gray sclera Increased bruising Joint hypermobility Possible scoliosis Possible dentinogenesis imperfecta/opalescent dentin Conductive hearing loss in up to 50% Autosomal dominant

22
Osteogenesis Imperfecta
gray/blue sclera

23
Dentinogenesis imperfecta
Opalescent dentin/ Dentinogenesis imperfecta

24
OI Type IV (with or without opalescent dentin)
Osteoporosis and bone fragility without significantly blue/gray sclerae after birth or early onset deafness 25% have fractures at birth while in some cases fractures do not occur until later in life May be clinically confused with idiopathic juvenile Short stature Mild to moderate bone deformities Scoliosis Dentinogenesis imperfecta/opalescent dentin in some patients Possible hearing loss, but not as common as in type I Autosomal dominant

26
OI Type III Significant fractures at birth and/or infancy
Marked short stature Severe kyphoscoliosis with progressive platyspondyly Severe long bone progressive deformities, often require wheelchair Decreased muscle mass in arms and legs Loose joints Whites of the eyes (sclera) have a blue, purple, or gray tint Brittle teeth Possible hearing loss Possible cardiopulmonary problems Most autosomal dominant, ? Autosomal recessive types ? Additional loci
 have a blue, purple, or gray tint. Brittle teeth. Possible hearing loss. Possible cardiopulmonary problems. Most autosomal dominant, Autosomal recessive types. Additional loci.")
28
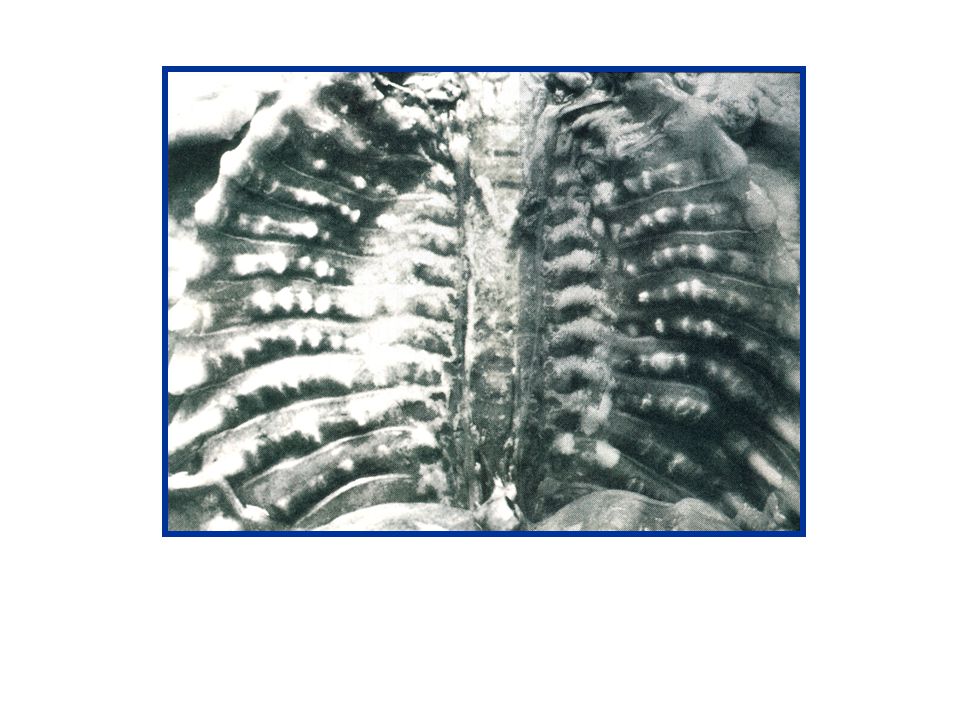
OI Type II Most severe form with extreme bone fragility at birth
Continuous beaded ribs with crumpled long bones Crumpled long bones with few rib fractures Narrow, dysplastic beaded ribs with poorly molded long bones and fractures Frequently lethal at or shortly after birth Numerous fractures Severe bone deformities Very small stature Underdeveloped lungs Abnormal brain neuropathology Diminished and structurally abnormal type I collagen Most cases autosomal dominant mutations with germline mosaicism

30
Skeletal dysplasia classified according to the region and type of bone involved
Predominantly epiphyseal Multiple epiphyseal dysplasia Hereditary arthro-ophthalmopathy (Stickler syndrome) Chondrodysplasia punctata Predominantly metaphyseal Lethal short limb dwarfism Achondroplasia Metaphyseal chondrodysplasias Major involvement of the spine Spondyloepiphyseal dysplasia ‘congenita’ SED (x-linked and others) Pseudoachondroplasia Kneist syndrome Diastrophic dwarfism Predominant involvement of single sites e.g. mesomelic dwarfism Abnormal bone density and/or modelling defects e.g. sclerosing disorders of bone From: Pope FM, Smith R. Color Atlas of Inherited Connective Tissue Disorders, Mosby-Wolfe, 1995
 Chondrodysplasia punctata. Predominantly metaphyseal. Lethal short limb dwarfism. Achondroplasia. Metaphyseal chondrodysplasias. Major involvement of the spine. Spondyloepiphyseal dysplasia ‘congenita’ SED (x-linked and others) Pseudoachondroplasia. Kneist syndrome. Diastrophic dwarfism. Predominant involvement of single sites. e.g. mesomelic dwarfism. Abnormal bone density and/or modelling defects. e.g. sclerosing disorders of bone. From: Pope FM, Smith R. Color Atlas of Inherited Connective Tissue Disorders, Mosby-Wolfe,")
32
OI - Autosomal Dominant
If a child inherits an altered collagen allele, the child will have the same type of OI as the parent Parent with OI Unaffected Parent Child with OI = Normal Gene = altered Gene

33
‘Protein suicide’ Normal typical Type I typical type II okay degraded

34
How is OI inherited when there is no family history of OI?
5-8% of families without a prior history of OI have multiple children with OI Due to “new” or “spontaneous” alteration occurs in the gonadal cells which give rise to multiple sperm or eggs. Germline or gonadal mosaicism - parent not affected by the condition, but mutation in certain percentage of his or her reproductive/germline cells

35
Clinical Testing Diagnostic confirmation of type
Two available tests for OI Protein/Biochemical DNA Prenatal diagnosis available using ultrasound (OI type II), amniocentesis or chorionic villus sampling.
, amniocentesis or chorionic villus sampling.")
36
How is OI treated? Bisphosphates, calcium, and Vitamin D
Prevent/control symptoms, maximize mobility, and developing optimal bone mass and muscle strength. Physical therapy, exercise, maintain normal weight Proper care of fractures Activity guidance - avoid high risk of injury activities Pain management Surgical placement of metal rods in the bones to provide strength Motility aids, such as wheelchairs and braces Avoid smoking, excessive alcohol, steroids Regular dental care Bisphosphates, calcium, and Vitamin D

37
Ehlers-Danlos Syndrome(s)
1 in 5, ,000 people both males and females all racial and ethnic backgrounds

38
Skin: soft velvety; hyperextensible; fragile - tears or bruises easily; severe parchment paper scars; slow and poor wound healing; molluscoid pseudotumors over pressure areas, striae Joints: hypermobile/hyperextensible, loose/unstable; prone to dislocations and/or subluxations; pain; hyperextensible joints; early osteoarthritis; chronic, early onset, debilitating musculoskeletal pain, pes planus, scoliosis Other: mitral valve prolapse; blue sclerae, gum disease, organ and vascular fragility

39
Six types of Ehlers-Danlos syndrome
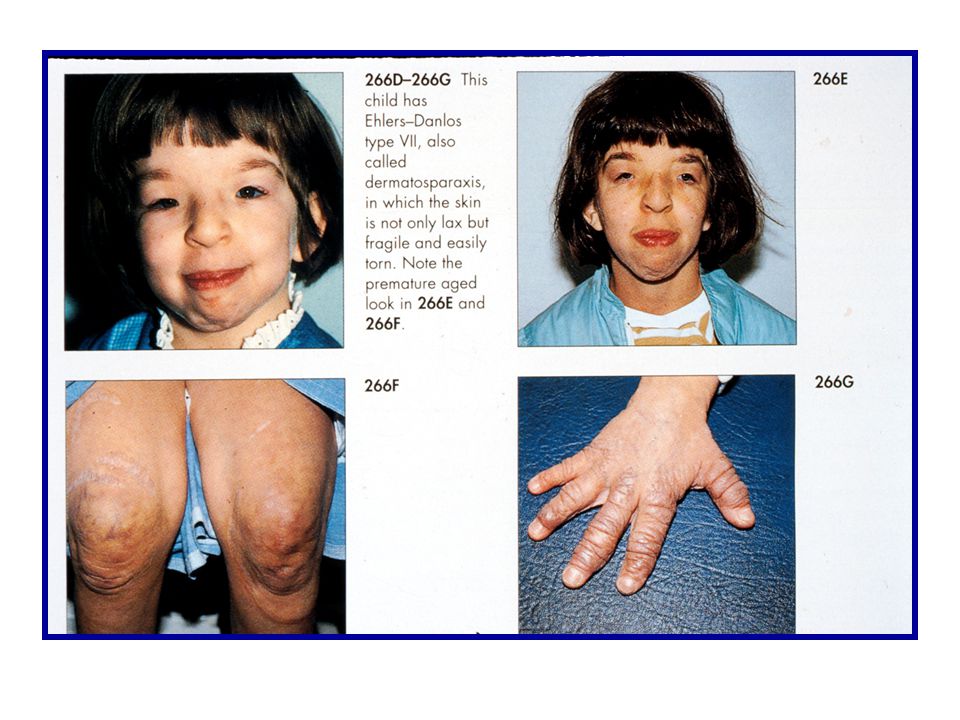
Classic Type (types I and II) AD - Type V and type ? collagen alterations “Parchment paper” scars, joint hypermobility,skin hyperelasticity, bruising Hypermobility Type (type III) AD - Type ? collagen alterations Joint hypermobility/extensibility, joint pain, premature arthritis Vascular Type (type IV) AD - Type III collagen alterations Arterial/intestinal/uterine fragility or rupture, bruising, translucent skin Kyphoscoliosis Type (type VI) AR - Lysyl hydroxylase deficiency Scoliosis at birth, scleral fragility Arthrochalasia Type (types VII A, VII B) AD - Type I collagen alterations Hypotonia, congenital hip dislocation Dermatosparaxis Type (type VII C) AR - Amino proteinase deficiency Severe skin fragility, doughy skin, blue sclerae, short stature, severe bruising
 AD - Type V and type collagen alterations. Parchment paper scars, joint hypermobility,skin hyperelasticity, bruising. Hypermobility Type (type III) AD - Type collagen alterations. Joint hypermobility/extensibility, joint pain, premature arthritis. Vascular Type (type IV) AD - Type III collagen alterations. Arterial/intestinal/uterine fragility or rupture, bruising, translucent skin. Kyphoscoliosis Type (type VI) AR - Lysyl hydroxylase deficiency. Scoliosis at birth, scleral fragility. Arthrochalasia Type (types VII A, VII B) AD - Type I collagen alterations. Hypotonia, congenital hip dislocation. Dermatosparaxis Type (type VII C) AR - Amino proteinase deficiency. Severe skin fragility, doughy skin, blue sclerae, short stature, severe bruising.")
40
Diagnostic Criteria for Classic EDS (I and II)
Autosomal Dominant Major Criteria Skin hyperextensibility Widened atrophic scars Joint hypermobility Minor Criteria Smooth, velvety skin Molluscoid pseudotumors Subcutaneous spheroids Joint complications (eg. sprains, dislocations, subluxations, pes planus) Muscle hypotonia, gross motor delays Easy bruising Complications of fragile tissue (hiatal hernia, anal prolapse) Surgical complications (postoperative hernias) Positive family history Laboratory Diagnosis Type V collagen abnormalities ( < 50%, most unknown) EM with ‘cauliflower’ fibrils
 Muscle hypotonia, gross motor delays. Easy bruising. Complications of fragile tissue (hiatal hernia, anal prolapse) Surgical complications (postoperative hernias) Positive family history. Laboratory Diagnosis. Type V collagen abnormalities ( < 50%, most unknown) EM with ‘cauliflower’ fibrils.")
42
Diagnostic Criteria for Hypermobility Type EDS
Autosomal Dominant Major Criteria Generalized joint hypermobility Skin hyperextensibility OR smooth, velvety skin (if atrophic scars are present, probably classic type) Minor Criteria Recurring joint dislocations (frequently shoulder, patella, and TMJ) Chronic joint/limb pain - often early onset and possibly debilitating Positive family history Laboratory Diagnosis None Distinguish from familial hypermobility (?)
 Minor Criteria. Recurring joint dislocations (frequently shoulder, patella, and TMJ) Chronic joint/limb pain - often early onset and possibly debilitating. Positive family history. Laboratory Diagnosis. None. Distinguish from familial hypermobility ( )")
43
Hypermobility EDS

44
Diagnostic Criteria for Vascular EDS
Autosomal Dominant Major Criteria: Two major criteria - highly indicative of the diagnosis, One - be very suspicious especially if family history Thin, translucent skin Arterial/intestinal/uterine fragility or rupture Extensive bruising Characteristic facial appearance with decreased subcutaneous adipose tissue, particularly in face and limbs, often with big eyes, pinched nose Minor Criteria: One or more minor criteria with positive family history, be suspicious Acrogeria Hypermobility of small joints Tendon and muscle rupture Talipes equinovarus Early onset varicose veins Arteriovenous, carotid-cavernous sinus fistula Pneumothorax/pneumohemothorax Gingival recession Positive family history or vascular or organ rupture, sudden early death in relatives Laboratory Diagnosis: Type III collagen abnormalities in fibroblasts, COL3A1 mutation

45
Blood dissects along the media (asterisks).
Compressed carotid artery by blood dissecting upward from a dissection. Blood dissects along the media (asterisks). * Translucent skin
. * Translucent skin.")
46
Confirmed by DNA testing 3 16 15 14
31 90s Ruptured renal artery “Marfan disease” 35 32 43 17 Vertebral aneurysm Spontaneous pneumothorax Sigmoidal perforation Ruptured thoracic artery Unaffected Probable EDS IV EDS IV- Vascular type Confirmed by DNA testing 3 16 15 14 Bilateral inguinal hernias Easy bruising Soft, translucent skin Increased bleeding

47
Type III Collagen Consists of 3 a1(III) chains (COL3A1 on chr 2)
Thin collagen fibers created Forms a loosely woven meshwork of reticular fibers Largely in: skin (with type I and type V collagens) blood vessels internal organs (e.g. smooth muscle layers GI tract, uterus, bladder)
 blood vessels. internal organs (e.g. smooth muscle layers GI tract, uterus, bladder)")
48
Vascular EDS Manifestations (Pepin et al, NEJM, 2000)
220 patients, 199 relatives Mean age at ascertainment: 28.7 Patients: (M , F ); Relatives: Family history present: Yes: % No: 41.4% Age of first complication in index patients (n=136) (M , F ) 25% had significant complication before 20 years 80% had significant complications before 40 years Age at death ranged from years, median = 48
; Relatives: Family history present: Yes: 38.2 % No: 41.4% Age of first complication in index patients (n=136) (M , F ) 25% had significant complication before 20 years. 80% had significant complications before 40 years. Age at death ranged from years, median = 48.")
49
Vascular EDS Manifestations (Pepin et al, NEJM, 2000)
Type of first complication in index patients

50
Vascular EDS Manifestations (Pepin et al, NEJM, 2000)
Causes of death (n =131)
")
51
Vascular EDS Manifestations (Pepin et al, NEJM, 2000)
Cause of pregnancy related deaths in 12 of 81 women (167 live births) Maternal death rate 1 in 23 (11.5%) Arterial rupture at delivery Uterine rupture during labor Arterial rupture in postpartum period
 Maternal death rate 1 in 23 (11.5%) Arterial rupture at delivery. Uterine rupture during labor. Arterial rupture in postpartum period.")
52
Treatment/Management of Vascular EDS
No specific therapies delay onset of complications. No proven medical surveillance for complications of Vascular EDS. The diagnosis should be established to provide genetic counseling, anticipatory guidance and education, and appropriate management strategies for surgeries and pregnancies.

53
Defect in N-Proteinase

54
Diagnostic Criteria for Dermatosparaxis Type EDS
Autosomal recessive Major Criteria Severe skin fragility Sagging redundant skin Minor Criteria Soft, doughy skin texture Easy bruising Premature rupture of fetal membranes Large hernias Laboratory diagnosis Biochemical confirmation of procollagen N-terminal peptidase (N-proteinase) by analysis of pNa1(I) and pNa2(I) in fibroblasts Determine level of N-proteinase activity
 by analysis of pNa1(I) and pNa2(I) in fibroblasts. Determine level of N-proteinase activity.")
56
Patient 1 Patient 2 Normal
Dermatosparaxis

57
Genetics of Ehlers-Danlos syndrome
Classic Type (types I and II) AD - Type V and type ? collagen alterations Vascular Type (type IV) AD - Type III collagen alterations Dermatosparaxis Type (type VII C) AR - Amino proteinase deficiency
 AD - Type V and type collagen alterations. Vascular Type (type IV) AD - Type III collagen alterations. Dermatosparaxis Type (type VII C) AR - Amino proteinase deficiency.")
58
Treatment/Management of EDS
Joint care Skin care Medications: Pain medications (avoid excessive NSAIDS in vascular type) Ascorbic acid (vitamin C) g/day in adults, 0.5 to 1.0 mg/day in children Consider risks/benefits of anticoagulation in vascular type Medical and Surgical Care: Be sure all health care providers know about the diagnosis (medic alert tag) Obtain emergent care for any new onset of pain and/or neurological systems Proceed with caution - tissue fragility leads to tears, excessive bleeding Manage hypertension, avoid constipation, carefully consider risks of pregnancy Lifestyle: Avoid contact sports and other high risk activities Maintain ideal body weight
 Ascorbic acid (vitamin C) g/day in adults, 0.5 to 1.0 mg/day in children. Consider risks/benefits of anticoagulation in vascular type. Medical and Surgical Care: Be sure all health care providers know about the diagnosis (medic alert tag) Obtain emergent care for any new onset of pain and/or neurological systems. Proceed with caution - tissue fragility leads to tears, excessive bleeding. Manage hypertension, avoid constipation, carefully consider risks of pregnancy. Lifestyle: Avoid contact sports and other high risk activities. Maintain ideal body weight.")
59
Collagen Disorders - Summary
Collagen - main structural protein in body Many genetically distinct types of collagen all have triple helix structure polypeptide chains with large (Gly -X-Y)n domain post translational modifications and extracellular processing critical Type I collagen: Heterotrimer abundant in skin, bones and ligaments. mutations in associated genes cause autosomal dominant OI point mutations in glycine can causing structural collagen abnormality can be more severe than haploinsufficincy (protien suicide) multiple severely affected children with normal parent can arise due to germline mosaicism Type III collagen: Homotrimer, mutations in gene encoding tyope III collagen associated with Vascular EDS (type IV EDS) characterized by vascular and tissue fragility Many types of EDS but classic EDS usually has skin and joint manifestations
n domain. post translational modifications and extracellular processing critical. Type I collagen: Heterotrimer. abundant in skin, bones and ligaments. mutations in associated genes cause autosomal dominant OI. point mutations in glycine can causing structural collagen abnormality can be more severe than haploinsufficincy (protien suicide) multiple severely affected children with normal parent can arise due to germline mosaicism. Type III collagen: Homotrimer, mutations in gene encoding tyope III collagen associated with Vascular EDS (type IV EDS) characterized by vascular and tissue fragility. Many types of EDS but classic EDS usually has skin and joint manifestations.")
Similar presentations













 keratin, myosin, fibrinogen - mainly alpha helix.>")


>")




 “Brittle bone disease”>")



