Download presentation
Presentation is loading. Please wait.

1
Nagi ALHaj MOLECULAR DIAGNOSIS OF GENETIC EPILEPSY
Prof of Molecular Biology Assist, Faculty Medicine , Sana’a ,University, Yemen Consultant of Genetic Center 48 MH

2
Overview on Genetic Epilepsy
A genetic contribution to etiology has been estimated to be present in about 40% of patients with epilepsy.. Three major groups: Mendelian disorders, in which a single major locus can account for segregation of the disease trait Non-mendelian or 'complex' diseases, in which the pattern of familial clustering can be accounted for by the interaction of the maternal inheritance pattern of mitochondrial DNA Chromosomal disorders, in which a gross cytogenetic abnormality is present.

3
The ‘common’ non-familial idiopathic epilepsies tend to display ‘complex’ inheritance.
They including various forms of Idiopathic generalised epilepsy (IGE) Juvenile myoclonic epilepsy (JME) Childhood absence epilepsy (CAE)
 Juvenile myoclonic epilepsy (JME) Childhood absence epilepsy (CAE)")
4
Genetics and Mutation Mutations in over 2,000 genes have now been identified in patients with more than 3,000 different disease phenotypes. For the clinicians and their patients, it is becoming increasingly important to obtain a genetic diagnosis Identifying the genetic aetiology of a disease may influence clinical management and will provide information regarding risk to future pregnancies. With the advent of high-throughput capillary sequencers and sequence analysis software, direct sequencing provides an accurate method for single gene analysis.

5
The inheritance pattern can be autosomal dominant, autosomal recessive, or X-linked. Mutations in a single gene may be associated with different types of seizures (clinical heterogeneity), and, conversely, mutations in different genes can cause the same epilepsy phenotype (genetic heterogeneity).
, and, conversely, mutations in different genes can cause the same epilepsy phenotype (genetic heterogeneity).")
6
Traditional nomenclature of inherited epilepsy:
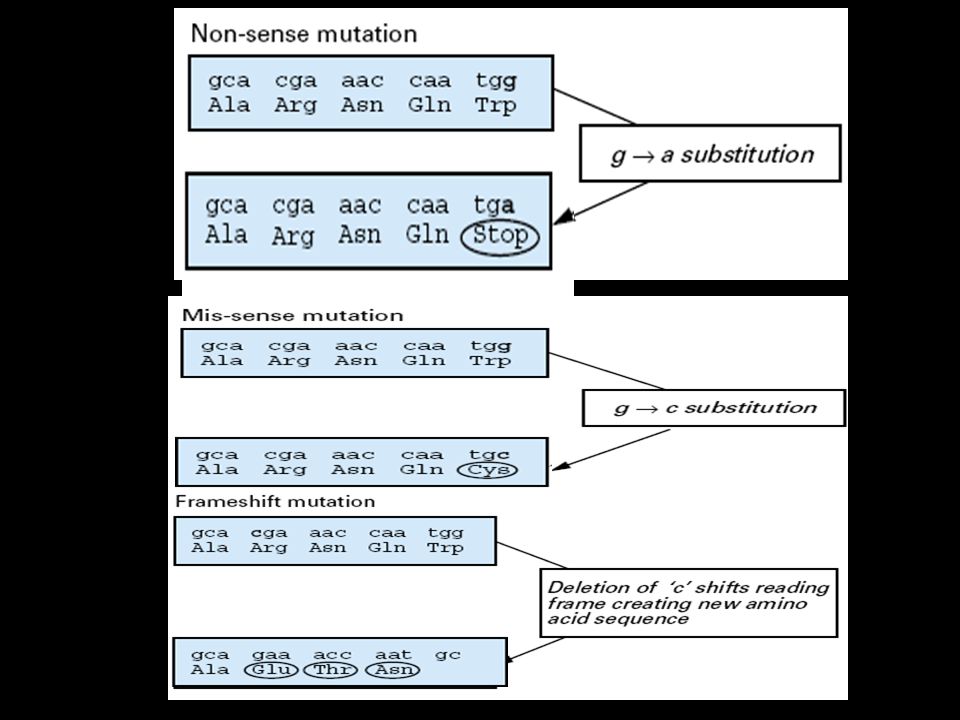
Different mutations in different genes can result in different phenotypes Different mutations in different genes can result in similar phenotypes Different mutations within one gene can result in different phenotypes An identical mutation within one gene can result in different phenotypes in different individuals (cause: environment, other genes)
")
7
Mutation identification begins with a
phenotype and proceeds toward the genotype genotype phenotype

10
The diagram shows the distribution of all genetic differences that had been mapped to chromosome 1 at the time this diagram was drawn.

11
Mutation identification by linkage analysis
Mutational analysis can be used to identify cells or DNA that have genotype and allele frequency differences from the normal genome. mutation site Genome scan has been replaced by mutational analysis but in a small number of families in whom the mutation cannot be identified Remains the only method for the genetic diagnosis of carriers.

12
Mutant alleles generating defects in particular proteins could disrupt the dance .
Cells homozygous for a mutant allele might be unable to complete chromosome duplication or mitosis or cytokinesis because a required component of the molecular machinery is missing or unable to function. A genetic map of part of the human X chromosome.

13
disease mechanism genotype phenotype
Elucidation of a disease mechanism presents a much more complex set of challenges cell protein network disease mechanism mRNA genotype phenotype

14
disease mechanism genotype phenotype
Not all potential defects arise from each mutation The number of potential defects increases exponentially with each emergent stage of complexity cell protein network disease mechanism mRNA genotype phenotype

15
disease mechanism T genotype phenotype
Not all potential defects arise from each mutation The most effective target for therapy ( ) would be the DNA mutation, but this is currently unfeasible T cell protein network disease mechanism mRNA T genotype phenotype
 would be. the DNA mutation, but this is currently unfeasible. T. cell. protein. network. disease. mechanism. mRNA. T. genotype. phenotype.")
16
T disease mechanism T genotype phenotype
The most effective target for therapy ( ) would be the DNA mutation, but this is currently unfeasible T T Proteins are also excellent targets for intervention cell protein network disease mechanism mRNA T genotype phenotype
 would be. the DNA mutation, but this is currently unfeasible. T. T. Proteins are also excellent targets for intervention. cell. protein. network. disease. mechanism. mRNA. T. genotype. phenotype.")
17
The genetic contribution to epilepsy: the known and missing heritability.
The Causes of Epilepsy, eds S.D. Shorvon et al, pp 6367. Cambridge University Press, Cambridge, 2011).
.")
18
Genes and mutations in neonatal syndromes and GEFS+
KCNQ2 Chr. 20q13.3 Benign familial neonatal convulsions (BFNC); Benign neonatal epilepsy-1 (EBN1) BFNC/myokymia syndrome KCNQ3 Chr. 8q24 Benign familial neonatal convulsions (BFNC); Benign neonatal epilepsy-2 (EBN2)
; Benign neonatal epilepsy-1 (EBN1) BFNC/myokymia syndrome. KCNQ3. Chr. 8q24. Benign familial neonatal convulsions (BFNC); Benign neonatal epilepsy-2 (EBN2)")
19
SCN1B SCN1A GABRG2 SCN2A Chr. 19q13
Generalized epilepsy with febrile seizures plus, type 1 (GEFS+ type 1; GEFSP1) SCN1A Chr. 2q24 GEFS+ type 2; GEFSP2 Severe myoclonic epilepsy in infancy (SMEI) GABRG2 Chr. 5q33-q34 GEFS+ type 3; GEFSP3 SMEI SCN2A Chr. 2q23-q24 Febrile seizures associated with afebrile seizures Benign familial neonatal-infantile seizures (BFNIS)
 SCN1A. Chr. 2q24. GEFS+ type 2; GEFSP2. Severe myoclonic epilepsy in infancy (SMEI) GABRG2. Chr. 5q33-q34. GEFS+ type 3; GEFSP3. SMEI. SCN2A. Chr. 2q23-q24. Febrile seizures associated with afebrile seizures. Benign familial neonatal-infantile seizures (BFNIS)")
20
Unknown Unknown Unknown Unknown Chr. 19q12-q13.1
Benign familial infantile convulsions, type 1 (BFIS type 1; BFIC1) Unknown Chr. 16p12-q12 BFIS type 2; BFIC2 Infantile convulsions and paroxysmal choreoathetosis (ICCA) Paroxysmal kinesigenic choreoathetosis (PKC) Unknown Chr. 16p12-p11.2 Rolandic epilepsy, paroxysmal exercise-induced dystonia, writer's cramp (RE-PED-WC) Unknown Chr. 16p13 Autosomal recessive (familial) benign idiopathic myoclonic epilepsy of infancy (FIME)
 Unknown. Chr. 16p12-q12. BFIS type 2; BFIC2. Infantile convulsions and paroxysmal choreoathetosis (ICCA) Paroxysmal kinesigenic choreoathetosis (PKC) Unknown. Chr. 16p12-p11.2. Rolandic epilepsy, paroxysmal exercise-induced dystonia, writer s cramp (RE-PED-WC) Unknown. Chr. 16p13. Autosomal recessive (familial) benign idiopathic myoclonic epilepsy of infancy (FIME)")
21
Traditional nomenclature of inherited epilepsy:
Gene 1 Gene 2 Gene 3 Gene 4 Mutations: Phenotype: B A C D F,G E Syndrome “A”: Gene 1 Gene 2 Gene 3 Gene 4 Syndrome “B”: Gene 2 Gene 1 Syndrome “C”: Gene 3 Gene 1 Syndrome “D”: Gene 1 Gene 4 Gene 2 Gene 3 Syndrome “E”: Syndrome “F”: Syndrome “G”: etc…

22
Gene-centric nomenclature of inherited epilepsy:
Mutations: Phenotype: B A C A D B A E F,G A C C E E A Gene 1: Phenotypic range A, B, C, D Gene 2: Phenotypic range A, B, E, F, G Gene 3: Phenotypic range A, S, E Gene 4: Phenotypic range A, E etc… Both “Traditional” and “Gene-centric” nomenclatures have specific advantages and disadvantages

23
Molecular Diagnosis

24
PATIENT HISTORY FOR MOLECULAR GENETIC TESTING

25
(1) The Infantile Epilepsy includes sequencing and deletion/duplication analysis of 38 genes causing Mendelian forms of epilepsy with onset of seizures during the first year of life. 38 gene activity, including voltage-gated sodium channels, the voltage-gated calcium channels, and gamma-aminobutyric acid (GABAA) receptors.
 receptors.")
27
(2) The Childhood-Onset Epilepsy 40 Genes (3)The Adolescent-Onset Epilepsy 21 Genes
Includes sequencing and deletion/duplication analysis of 40 and 21 genes respectively that causing Mendelian forms of epilepsy. Genes that encode nicotinic acetylcholine receptors and calcium channels,

29
Methodology Infantile Epilepsy Panel The Childhood-Onset Epilepsy
The Adolescent-Onset Epilepsy Using genomic DNA obtained from blood, ~ 570 coding exons and the flanking splice junctions of 38 genes are sequenced simultaneously by (next-generation sequencing). The sequence is assembled and compared to published genomic reference sequences. Sanger sequencing is used to compensate for low coverage and refractory amplifications in regions where pathogenic mutations have been previously published.
. The sequence is assembled and compared to published genomic reference sequences. Sanger sequencing is used to compensate for low coverage and refractory amplifications in regions where pathogenic mutations have been previously published.")
30
Individual nucleotides
DNA in the Cell chromosome cell nucleus Double stranded DNA molecule Individual nucleotides Target Region for PCR

31
Extraction of DNA from whole Blood
Extract and discard plasma, taking care not to remove the buffy coat.

32
Genetic Lab 48 MH

33
DNA Amplification with the Polymerase Chain Reaction (PCR)
Separate strands (denature) 5’ 3’ Starting DNA Template 5’ 3’ Add primers (anneal) 5’ 3’ Forward primer Reverse primer 5’ 3’ Make copies (extend primers)
 5’ 3’ Starting DNA Template. 5’ 3’ Add primers (anneal) 5’ 3’ Forward primer. Reverse primer. 5’ 3’ Make copies (extend primers)")
34
PCR Copies DNA Exponentially through Multiple Thermal Cycles
Original DNA target region Thermal cycle Thermal cycle Thermal cycle In 32 cycles at 100% efficiency, 1.07 billion copies of targeted DNA region are created

35
Deletion analysis of the SCN1A gene by multiplex PCR.
loss of bands loss of bands Deletion analysis of the SCN1A gene by multiplex PCR.

36
COMPARATIVE GENOMIC HYBRIDIZATION (CGH)
")
37
Identification of a deletion encompassing gene in a male patient.
Y-axes represent R Log ratio B allele frequency The red line (log R ratio profile) corresponds to the median smoothing series. The X-axis indicates the position on the X chromosome Identification of a hemizygous Xq22.1 deletion with a 370 K SNP microarray
 corresponds to the median smoothing series. The X-axis indicates the position on the X chromosome. Identification of a hemizygous Xq22.1 deletion with a 370 K SNP microarray.")
38
Figure 1. Identification of a deletion encompassing PCDH19 in a male patient.
Black horizontal bars represent the gene PCDH19) and pseudogenes comprised in the deleted region Analysis of the patient and his mother with CGH microarrays, showing that the new deletion occurred.
 and pseudogenes comprised in the deleted region. Analysis of the patient and his mother with CGH microarrays, showing that the new deletion occurred.")
40
ABI Prism 310 Genetic Analyzer

41
3’-TAAATGATTCC-5’ A AT ATT ATTT ATTTA ATTTAC ATTTACT ATTTACTA
ATTTACTAA ATTTACT ATTTAC ATTT ATTTA AT ATTTACTA ATTTACTAAG ATTTACTAAGG A DNA template 5’ 3’ Primer anneals Extension produces a series of ddNTP terminated products each one base different in length Each ddNTP is labeled with a different color fluorescent dye Figure 10.5 DNA sequencing process with fluorescent ddNTPs. A primer that has been designed to recognize a specific region of a DNA template anneals and is extended with a polymerase. Because a mixture of dNTPs and ddNTPs exist for each of the four possible nucleotides, some of the extension products are halted by incorporation of a ddNTP while other molecules continue to be extended. Each ddNTP is labeled with a different dye that enables each extension product to be distinguished by color. A size-based separation of the extension products permits the DNA sequence to be read provided that sufficient resolution is present to clearly see each base.

42
Epilepsy GEFS

43
OMIM Record Link to Gene Epilepsy GEFS Coriell Cell Repositories
Human Gene Mutation Database

44
Gene Links to Everywhere (almost) Epilepsy GEFS
 Epilepsy GEFS")
45
Gene GEFS Genome Maps GEFS NM records NT Gene Model UniGene Gene

46
Detection of 9 different point mutations of PCDH19 in 11 female patients by direct sequencing.
The A of the ATG translation initiation codon in the reference sequence Sequence electropherograms of the mutations and the missense variant (c.3319C>G/p.Arg1107Gly) identified in association with the c.859G>T/p.Glu287X nonsense mutation.
 identified in association with the c.859G>T/p.Glu287X nonsense mutation.")
47
Alignment of the regions surrounding the mutations (indicated by an arrow) in orthologous and paralogous proteins, showing the high conservation of each affected amino-acid in vertebrates and in the delta protocadherin paralogous genes.

48
FISH analysis of the gene deletion in the male patient showing somatic mosaicism in fibroblasts.
A) Absence of the specific Xq22.1 probe site on metaphase chromosomes (PBL); (B) In fibroblasts, presence of one hybridization spot in 53% of the cells and absence of signal in the remaining 47%; FISH analysis on PBL (C) and fibroblasts (D) of a female control. PCDH19-specific signals (red) are indicated by arrowheads.
 Absence of the specific Xq22.1 probe site on metaphase chromosomes (PBL); (B) In fibroblasts, presence of one hybridization spot in 53% of the cells and absence of signal in the remaining 47%; FISH analysis on PBL (C) and fibroblasts (D) of a female control. PCDH19-specific signals (red) are indicated by arrowheads.")
49
THANK YOU

Similar presentations












 Polymerase Chain Reaction (PCR) Microarray analysis Link to Gene Therapy.>")










