Download presentation
Presentation is loading. Please wait.

1
Addison’s Disease Nasimeh Rakhshani CC3

2
Adrenal Insufficiency
Addison’s Disease

3
Addison’s Disease First described by Thomas Addison in 1855
His description referred to primary adrenal insufficiency At that time, the most common etiology was tuberculosis infiltration TB infiltration continues to be the leading cause worldwide

4
Adrenal Insufficiency (AI)
Impairment in synthesis and/or release of adrenocortical hormones Classified as: Primary AI results from disease intrinsic to the adrenal cortex Secondary AI results from impaired release or effect of adrenocorticotropic hormone (ACTH) from the pituitary gland Tertiary AI results from the impaired release or effect of corticotropin releasing hormone (CRH) from the hypothalamus
 from the pituitary gland. Tertiary AI results from the impaired release or effect of corticotropin releasing hormone (CRH) from the hypothalamus.")
5
The HPA Axis (-) Hypothalamus CRH (-) ACTH Glucocorticoids
Adrenal Gland

6
Clinical Manifestations
In general, symptoms of AI include fatigue and GI complaints (nausea and vomiting) Clinical suspicion is important because the presentation of AI may be insidious and subtle Resultantly, clinical diagnosis is frequently delayed or missed early if unrecognized, may present in a life-threatening crisis with acute cardiovascular collapse (adrenal crisis) Simm PJ et al (2004), of found in children diagnosed with primary adrenal insufficiency that there was a median of 2 years delay between the onset of the first symptoms and the diagnosis
 Clinical suspicion is important because the presentation of AI may be insidious and subtle. Resultantly, clinical diagnosis is frequently delayed or missed early if unrecognized, may present in a life-threatening crisis with acute cardiovascular collapse (adrenal crisis) Simm PJ et al (2004), of found in children diagnosed with primary adrenal insufficiency that there was a median of 2 years delay between the onset of the first symptoms and the diagnosis.")
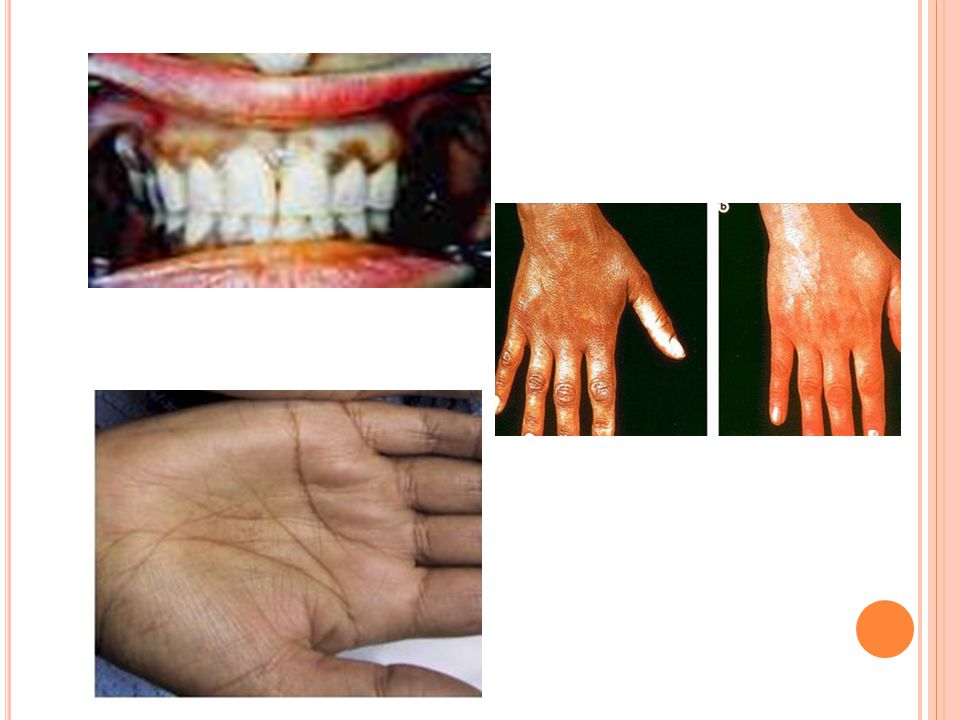
7
Clinical Manifestations
Signs and symptoms of primary adrenal insufficiency vary depending on which hormones are deficient and the severity of the defects

8
Clinical Manifestations cont’d
Glucocorticoid Deficiency Mineralocorticoid Deficiency Adrenal Androgen Deficiency Fasting hypoglycemia Hypotension Decreased axillary hair (females) Increased insulin sensitivity Dizziness Decreased pubic hair (females) Muscle weakness Salt craving Loss of libido (females) Morning headache Weight loss If prepubescent: assymptomatic ↑ production of POMC ↑ melanin Anorexia ↑pigmentation: palmer creases, gingival border,axilla Electrolyte anomalies (hypoNa, hyperK, metabolic acidosis) Mineralocorticiod = aldosterone and symptoms mostly b/c of sodium loss Only females because in males androgen production is mostly by testes
 Increased insulin sensitivity. Dizziness. Decreased pubic hair (females) Muscle weakness. Salt craving. Loss of libido (females) Morning headache. Weight loss. If prepubescent: assymptomatic. ↑ production of POMC ↑ melanin. Anorexia. ↑pigmentation: palmer creases, gingival border,axilla. Electrolyte anomalies (hypoNa, hyperK, metabolic acidosis) Mineralocorticiod = aldosterone and symptoms mostly b/c of sodium loss. Only females because in males androgen production is mostly by testes.")
10
Clinical Manifestations
Adrenal Crisis Hypotension or shock Particularly if disproportionate to apparent underlying illness Serum electrolyte and metabolic abnormalities: Hyponatremia Hyperkalemia Metabolic Acidosis Hypoglycemia Vomiting and diarrhea, sometimes with severe abdominal pain Unexplained fever, weight loss and anorexia

11
Clinical Manifestations
Adrenal Crisis: When to be suspicious??? In neonates, presents within 1st few days-weeks of life with vomiting, diarrhea, ↓BP, ↓Na, ↑K and ↓BG CAH (21-hydroxylase deficiency) most common cause In females with CAH is suggested by ambiguous genitalia In infants and older children with previously diagnosed AI Weight loss, serum electrolyte abnormalities +/- hyperpigmentation Often a history of an antecedent precipitating stress (eg, surgery or infection) Bilateral adrenal hemorrhage or infarction Children with hypotension and shock that fail to respond to vigorous fluid resuscitation and inotropic medications Especially if have severe hyponatremia and hyperkalemia
 most common cause. In females with CAH is suggested by ambiguous genitalia. In infants and older children with previously diagnosed AI. Weight loss, serum electrolyte abnormalities +/- hyperpigmentation. Often a history of an antecedent precipitating stress (eg, surgery or infection) Bilateral adrenal hemorrhage or infarction. Children with hypotension and shock that fail to respond to vigorous fluid resuscitation and inotropic medications. Especially if have severe hyponatremia and hyperkalemia.")
12
Etiology Steroidogenesis disorders: Defects within the biosynthetic pathways of glucocorticoids +/- mineralocorticoids lead to impaired synthesis of cortisol and/or aldosterone There are also drugs that inhibit cortisol synthesis (aminoglutethimide, ketoconazole, and etomidate) Adrenal damage: Injury from factors extrinsic to the adrenal gland may impair adrenal function Abnormal adrenal development: A lack of normal adrenocortical cell differentiation may result in adrenal hypoplasia Adrenal unresponsiveness to ACTH: Defects in adrenal responsiveness to ACTH results in cortisol deficiency Peroxisomal disorders: Accumulation of abnormal very long chain fatty acids within peroxisomes which may lead to adrenal impairment Adrenal damage; (eg, infection, drugs, antibodies, and hemorrhage) Congenital adrenal hyperplasia: 73 percent. Almost all of these cases were because of CYP21A2 (21-hydroxylase) deficiency Autoimmune adrenal failure: 13 percent Peroxisomal disorders: 5 percent Wolman disease (lysosomal acid lipase deficiency): 3 percent Adrenal hypoplasia congenita: 1 percent Triple A syndrome (Unresponsiveness to ACTH): 1 percent No diagnosis identified: 6 percent
 Adrenal damage: Injury from factors extrinsic to the adrenal gland may impair adrenal function. Abnormal adrenal development: A lack of normal adrenocortical cell differentiation may result in adrenal hypoplasia. Adrenal unresponsiveness to ACTH: Defects in adrenal responsiveness to ACTH results in cortisol deficiency. Peroxisomal disorders: Accumulation of abnormal very long chain fatty acids within peroxisomes which may lead to adrenal impairment. Adrenal damage; (eg, infection, drugs, antibodies, and hemorrhage) Congenital adrenal hyperplasia: 73 percent. Almost all of these cases were because of CYP21A2 (21-hydroxylase) deficiency. Autoimmune adrenal failure: 13 percent. Peroxisomal disorders: 5 percent. Wolman disease (lysosomal acid lipase deficiency): 3 percent. Adrenal hypoplasia congenita: 1 percent. Triple A syndrome (Unresponsiveness to ACTH): 1 percent. No diagnosis identified: 6 percent.")
13
Etiology Steroido-genesis disdorders Adrenal damage
Abnormal adrenal develop’t Unresponsive to ACTH Peroxisomal defects CAH Adrenal hemorrage X-linked AHC Familial GC deficiency X-linked ALD/AMN Defect in aldosterone production Infection (TB, HIV, fungal) Adrenal hypoplasia SF-1 defect - Allgrove syndrome (triple A syn) Neonatal ALD Defects in cholesterol biochemistry Autoimmune (polyglandular AIS) Refsum disease -Wolman dz Zellweger syndrome AIS autoimmune syndrome ALD adrenoleukodystrophy AMN adrenomyeloneuropathy adrenal hypoplasia congenita (AHC) Steroidogenic factor-1 (SF-1)
 Adrenal hypoplasia SF-1 defect. - Allgrove syndrome (triple A syn) Neonatal ALD. Defects in cholesterol biochemistry. Autoimmune (polyglandular AIS) Refsum disease. -Wolman dz. Zellweger syndrome. AIS autoimmune syndrome. ALD adrenoleukodystrophy AMN adrenomyeloneuropathy. adrenal hypoplasia congenita (AHC) Steroidogenic factor-1 (SF-1)")
14
Etiology Continued Perry et al (2005):
20 years of data, 103 patients <18 y/o Primary AI 73% congenital adrenal hyperplasia (CAH) 13% autoimmune adrenal insufficiency 5% Peroxisomal disorders 3% Wolman disease (lysosomal acid lipase deficiency) 1% Adrenal hypoplasia congenita 1% Triple A syndrome (Unresponsiveness to ACTH) 6% No diagnosis identified 1 in to recently reported their experience with primary adrenal insufficiency in children 18 years old in Montreal, Canada, over the past 20 years. Of the 103 patients The high false-positive rate, particularly in premature infants, has prevented universal adoption of biochemical screening for CAH.
 13% autoimmune adrenal insufficiency. 5% Peroxisomal disorders. 3% Wolman disease (lysosomal acid lipase deficiency) 1% Adrenal hypoplasia congenita. 1% Triple A syndrome (Unresponsiveness to ACTH) 6% No diagnosis identified. 1 in to recently reported their experience with primary adrenal. insufficiency in children 18 years old in Montreal, Canada, over the past 20 years. Of the 103 patients. The high false-positive rate, particularly in premature infants, has prevented universal adoption of biochemical screening for CAH.")
15
Diagnosis 3 Step Process:
Confirm adrenal insufficiency demonstrating inappropriately low cortisol secretion Determine whether the cortisol deficiency is primary or central AI Determine the cause of the underlying disorder

16
Diagnostic Tests Static Tests Dynamic Tests
Cortisol, ACTH, Adrenal Androgens Mineralocorticoid status Serum electrolytes Plasma renin activity (PRA), direct [renin] Dynamic Tests Short ACTH stimulation test: Serum cortisol levels are measured before and 60 minutes after the rapid IV infusion of synthetic ACTH (cosyntropin) Tests of ACTH secretory ability Insulin-induced hypoglycemia Glucagon Metyrapone test CRH stimulation test: helps determine if 2o or 3o AI PRA is a measure of the rate of conversion of angiotensinogen to angiotensin I [7], and direct renin measures the concentration of renin in the blood Cortisol secretion is deficient in patients with primary adrenal insufficiency despite the fact that their ability to secrete ACTH is intact. Conversely, patients with secondary or tertiary adrenal insufficiency have intrinsically normal but atrophic adrenal glands that are capable of producing cortisol but fail to do so because ACTH secretion is deficient. PLASMA RENIN ACTIVITY - Secondary adrenal insufficiency (ACTH deficiency) is diagnosed if there is a poor cortisol response to tests of ACTH secretory ability (in addition to low 8 a.m. cortisol and low ACTH). - Tertiary adrenal insufficiency (deficient secretion of CRH) is diagnosed if the tests of ACTH secretory ability are low (in addition to low 8 a.m. cortisol and low ACTH), and if ACTH levels rise in response to CRH. In the rare circumstance of a defective CRH receptor, there will be no CRH-stimulated rise in ACTH secretion. nsulin-induced hypoglycemia — This is the most sensitive standard test of ACTH release. Serial measurements of blood glucose and cortisol are made before and at 15, 30, 45, and 60 minutes after an infusion of insulin (0.05 units/kg). The peak cortisol level should be at least double the baseline level or greater than 20 mcg/dL [8]. If desired, samples for growth hormone can be obtained at the same time. A normal GH response is usually defined as ≥10 mcg/L. (See "Diagnosis of growth hormone deficiency in children", section on GH stimulation tests). Symptomatic hypoglycemia is desirable during the test, but marked changes in levels of consciousness or blood glucose <30 mg/dL must be treated with prompt administration of intravenous dextrose. The test is not recommended for children younger than 3 years old because of the risk of damage to the central nervous system resulting from hypoglycemia. Glucagon stimulation test — This test is more appropriate in children younger than 3 years. After an intramuscular injection of glucagon, the blood glucose level initially rises then drops rapidly because of the release of endogenous insulin. Serum cortisol and growth hormone levels should increase in response to this fall in blood glucose [9]. Metyrapone test — This test also is used in the assessment of ACTH secretory ability, but is rarely used clinically. Metyrapone blocks the activity of the enzyme 11-beta-hydroxylase, which is needed to convert 11-deoxycortisol to cortisol, causing a decrease in serum cortisol levels (show figure 2). In a normal patient, the decrease in cortisol levels will stimulate ACTH secretion and increase the production of cortisol precursors. This can be measured by a rise in serum levels of ACTH and 11-deoxycortisol and/or in urinary 17-hydroxycorticosteroids and free cortisol. The ACTH-stimulated gland eventually overrides the enzymatic block, allowing cortisol production to occur. However, patients must be observed closely during this test because acute adrenal insufficiency may be precipitated if the patient has marked ACTH deficiency. (See "Metyrapone stimulation tests")
, direct [renin] Dynamic Tests. Short ACTH stimulation test: Serum cortisol levels are measured before and 60 minutes after the rapid IV infusion of synthetic ACTH (cosyntropin) Tests of ACTH secretory ability. Insulin-induced hypoglycemia. Glucagon. Metyrapone test. CRH stimulation test: helps determine if 2o or 3o AI. PRA is a measure of the rate of conversion of angiotensinogen to angiotensin I [7], and direct renin measures the concentration of renin in the blood. Cortisol secretion is deficient in patients with primary adrenal insufficiency despite the fact that their ability to secrete ACTH is intact. Conversely, patients with secondary or tertiary adrenal insufficiency have intrinsically normal but atrophic adrenal glands that are capable of producing cortisol but fail to do so because ACTH secretion is deficient. PLASMA RENIN ACTIVITY. - Secondary adrenal insufficiency (ACTH deficiency) is diagnosed if there is a poor cortisol response to tests of ACTH secretory ability (in addition to low 8 a.m. cortisol and low ACTH). - Tertiary adrenal insufficiency (deficient secretion of CRH) is diagnosed if the tests of ACTH secretory ability are low (in addition to low 8 a.m. cortisol and low ACTH), and if ACTH levels rise in response to CRH. In the rare circumstance of a defective CRH receptor, there will be no CRH-stimulated rise in ACTH secretion. nsulin-induced hypoglycemia — This is the most sensitive standard test of ACTH release. Serial measurements of blood glucose and cortisol are made before and at 15, 30, 45, and 60 minutes after an infusion of insulin (0.05 units/kg). The peak cortisol level should be at least double the baseline level or greater than 20 mcg/dL [8]. If desired, samples for growth hormone can be obtained at the same time. A normal GH response is usually defined as ≥10 mcg/L. (See Diagnosis of growth hormone deficiency in children , section on GH stimulation tests). Symptomatic hypoglycemia is desirable during the test, but marked changes in levels of consciousness or blood glucose <30 mg/dL must be treated with prompt administration of intravenous dextrose. The test is not recommended for children younger than 3 years old because of the risk of damage to the central nervous system resulting from hypoglycemia. Glucagon stimulation test — This test is more appropriate in children younger than 3 years. After an intramuscular injection of glucagon, the blood glucose level initially rises then drops rapidly because of the release of endogenous insulin. Serum cortisol and growth hormone levels should increase in response to this fall in blood glucose [9]. Metyrapone test — This test also is used in the assessment of ACTH secretory ability, but is rarely used clinically. Metyrapone blocks the activity of the enzyme 11-beta-hydroxylase, which is needed to convert 11-deoxycortisol to cortisol, causing a decrease in serum cortisol levels (show figure 2). In a normal patient, the decrease in cortisol levels will stimulate ACTH secretion and increase the production of cortisol precursors. This can be measured by a rise in serum levels of ACTH and 11-deoxycortisol and/or in urinary 17-hydroxycorticosteroids and free cortisol. The ACTH-stimulated gland eventually overrides the enzymatic block, allowing cortisol production to occur. However, patients must be observed closely during this test because acute adrenal insufficiency may be precipitated if the patient has marked ACTH deficiency. (See Metyrapone stimulation tests )")
17
1. Confirming AI Initial step: measurement of serum ACTH and cortisol in the morning (8 a.m.) and in the fasting state If serum cortisol is low and serum ACTH is high likely primary AI and diagnosis can be confirmed with an ACTH stimulation test If low cortisol and normal lytes the rapid ACTH stimulation test often performed simultaneously If serum ACTH is also low, serum cortisol is indeterminate or pituitary disease is suspected likely central AI tests of ACTH secretory ability 8 am cort <80 nmol/l Following ACTH stim <500 nmol/l Normal ACTH 4.50

18
1. Confirming AI cont’d In patients suspected of adrenal crisis:
Immediate treatment crucial and must not postpone until diagnosis confirmed Draw required samples and initiate therapy (saline and GC replacement) Cortisol, ACTH, electrolytes, PRA, renin, etc. May perform a short ACTH stimulation test following treatment in these patients if: Treatment has been given for < a few days (no adrenal suppression secondary to treatment) Dexamethasone used to treat (not detected in cortisol assay unlike hydrocortisone and cortisone)
 Cortisol, ACTH, electrolytes, PRA, renin, etc. May perform a short ACTH stimulation test following treatment in these patients if: Treatment has been given for < a few days (no adrenal suppression secondary to treatment) Dexamethasone used to treat (not detected in cortisol assay unlike hydrocortisone and cortisone)")
19
2. Establish the level of defect
Diagnosis of Primary AI:↓ am cortisol and ↑ am ACTH levels + ↓ or absent cortisol in response to ACTH stimulation test Often evidence of mineralocorticoid deficiency hyponatremia, hyperkalemia, ↑ PRA and/or ↑ [renin] Diagnosis of Central AI: ↓ Basal and ACTH stimulated cortisol secretion and ↓ Basal ACTH tests of ACTH secretory ability +/- CRF stimulation test Practically, not always necessary to determine if AI is 2o or 3o treatment is often the same Plasma levels of renin and aldosterone are usually unaffected in central AI Plasma levels of renin and aldosterone are usually unaffected in secondary or tertiary adrenal insufficiency, but mineralocorticoid deficiency can sometimes occur after very prolonged deficiency of ACTH. So whether its primary vs. secondary or tertiary

20
3. Evaluation of Cause Primary AI Central AI
Evaluate for CAH (most common cause of 1o AI ) adrenal androgens Measure adrenal antibodies if +ive screen for autoimmune polyglandular syndromes Measuring antibodies to other endocrine glands (thyroid, parathyroid, and islet cell antibodies) If antibodies negative screen for other causes of 1o AI Imaging (CT) identifying adrenal hemorrhage, calcifications, or infiltrative disease Central AI Evaluate for secretion of other pituitary hormones Very long chain fatty acid panel
 adrenal androgens. Measure adrenal antibodies if +ive screen for autoimmune polyglandular syndromes. Measuring antibodies to other endocrine glands (thyroid, parathyroid, and islet cell antibodies) If antibodies negative screen for other causes of 1o AI. Imaging (CT) identifying adrenal hemorrhage, calcifications, or infiltrative disease. Central AI. Evaluate for secretion of other pituitary hormones. Very long chain fatty acid panel.")
21
Diagnostic Approach

22
Treatment of Adrenal Insufficiency
Principles: Maintenance Therapy (Replacement) Stressed Conditions
 Stressed Conditions.")
23
Treatment Cont’d Maintenance Therapy Glucocorticoid Therapy
Calculated according to body surface area Often hydrocortisone is preferred because of its short duration of action and low potency ease in titration to optimal dose (5-16 mg/m2/d divided into 3 doses) During follow-up must ensure adequate somatic growth (weight, BA, height and weight velocities) and screen for symptoms of insufficiency Must also screen for symptoms of GC excess Mineralocorticoid Therapy Fludrocortisone (Florinef) mg/d do not vary by age or surface area because aldosterone secretion rate is nearly constant throughout the lifespan Monitor for signs of inadequate replacement: dehydration, poor weight gain, salt-craving, and hyponatremia with hyperkalemia Routine measurement of plasma renin activity (PRA) is not generally useful in monitoring adequacy of treatment because levels of these hormones tend to be variable, based on posture and sodium intake, and are often above the normal range even in patients who are receiving adequate replacement therapy. However, on occasion, measurements of these hormones may be useful in monitoring compliance with therapy, or in response to an unexplained change in clinical status
 During follow-up must ensure adequate somatic growth (weight, BA, height and weight velocities) and screen for symptoms of insufficiency. Must also screen for symptoms of GC excess. Mineralocorticoid Therapy. Fludrocortisone (Florinef) mg/d do not vary by age or surface area because aldosterone secretion rate is nearly constant throughout the lifespan. Monitor for signs of inadequate replacement: dehydration, poor weight gain, salt-craving, and hyponatremia with hyperkalemia. Routine measurement of plasma renin activity (PRA) is not generally useful in monitoring adequacy of treatment because levels of these hormones tend to be variable, based on posture and sodium intake, and are often above the normal range even in patients who are receiving adequate replacement therapy. However, on occasion, measurements of these hormones may be useful in monitoring compliance with therapy, or in response to an unexplained change in clinical status.")
24
Treatment Cont’d Stress Conditions
Primary goal is to avoid serious consequences of an adrenal crisis always wear identification Illness: Minor stress (e.g. sore throat, rhinorrhea, T < 38ºC) may not require ∆ dose Moderate stress (e.g. severe URTI) double the GC replacement dose Major stress (e.g. T > 38ºC and/or vomiting), three to four times the GC replacement dose If child unable to keep down oral dose administer IM GC Surgery: During general anesthesia, +/- surgery, the GC requirements increases greatly Protocols vary depending on nature of surgery, length of surgery, age of patient etc Stress dosing is generally continued until the patient can tolerate oral intake, is afebrile, and is hemodynamically stable
 may not require ∆ dose. Moderate stress (e.g. severe URTI) double the GC replacement dose. Major stress (e.g. T > 38ºC and/or vomiting), three to four times the GC replacement dose. If child unable to keep down oral dose administer IM GC. Surgery: During general anesthesia, +/- surgery, the GC requirements increases greatly. Protocols vary depending on nature of surgery, length of surgery, age of patient etc. Stress dosing is generally continued until the patient can tolerate oral intake, is afebrile, and is hemodynamically stable.")
25
Thank You!!! Questions???

Similar presentations




, FRCP Consultant Endocrinologist>")


 By: Anna Heideman & Angela Mullins.>")




 are the triangle-shaped and orange- colored endocrine.>")






