Download presentation
Presentation is loading. Please wait.

1
Molecular Classification of Cancer: Class Discovery and Class Prediction by Gene Expression Monitoring Golub TR, Slonim DK, Tamayo P, Huard C, Gaasenbeek M, Mesirov JP, Coller H, Loh ML, Downing JR, Caligiuri MA, Bloomfield CD, Lander ES. Whitehead Institute/Massachusetts Institute of Technology Center for Genome Research, Cambridge, MA 02139, USA.

2
Contents Background Objective Methods Results and GeneCluster
Conclusion

3
Background: Cancer Classification
Cancer Classification has been primarily based on morphological appearance of tumor. Limitations of morphology classification: tumors of similar histopathological appearance can have significantly different clinical courses and response to therapy. Cancer classification has been difficult because it has historically relied on specific biological insights, rather than systematic unbiased approaches for recognizing tumor subtypes.

4
Background: Cancer Classification
No general approach has been made for identifying new cancer classes (class discovery) and assigning tumors to known classes(class prediction). A generic approach to cancer classification based on gene expression monitoring by DNA microarrays is described and applied to acute human leukemia as a test case.
 and assigning tumors to known classes(class prediction). A generic approach to cancer classification based on gene expression monitoring by DNA microarrays is described and applied to acute human leukemia as a test case.")
5
Background: Cancer Classification
Cancer classification can be divided into two challenges: class discovery and class prediction. Class discovery refers to defining previously unrecognized tumor subtypes. Class prediction refers to the assignment of particular tumor samples to already-defined classes. We will look at class prediction for classifying Acute Myeloid Leukemia (AML) and Acute Lymphoblastic Leukemia(ALL).
 and Acute Lymphoblastic Leukemia(ALL).")
6
Background:Leukemia Acute leukemia: Cancer of bone marrow.
Marrow cannot produce appropriate amount of red and white blood cells. Subtypes: acute Lymphoblastic leukemia (ALL) or acute myeloid leukemia (AML) Particular subtypes of acute leukemia have been found to be associated with specific chromosomal translocations. ALL : t(12;21)(p13;q22) in 25% of patients. AML: t(8;21)(q22;q22) in 15% of patients.
 or acute myeloid leukemia (AML) Particular subtypes of acute leukemia have been found to be associated with specific chromosomal translocations. ALL : t(12;21)(p13;q22) in 25% of patients. AML: t(8;21)(q22;q22) in 15% of patients.")
7
Background:Leukemia ALL : 58 % survival rate AML: 14% survival rate
Distinction between ALL and AML essential for successful treatment. Treatment: chemotherapy, bone marrow transplant. ALL : corticosteroids, vincristine, methotrexate, L-asparaginase AML :daunorubicin, cytarabine Although distinction between ALL and AML is well established, no single test is currently sufficient to establish the diagnosis, but a combination of different tests in morphology, histochemistry and immunophenotyping are performed in an specialized laboratory. Although usually accurate, leukemia classification remains imperfect and errors do occur.

8
Objective To develop a more systematic approach to cancer classification based on the simultaneous expression monitoring of thousands of genes using DNA microarrays with leukemia as test cases.

9
Overview Expression Data Known classes
Asses gene class correlation: Neighborhood Analysis Build Predictor Test Predictor by cross-validation Test Predictor on Independent Dataset

10
Method: Biological Samples
Primary samples: 38 bone marrow samples (27 ALL, 11 AML) obtained from acute leukemia patients at the time of diagnosis; Measured the expression of 6817 human genes using Affymetrix arrays.
 obtained from acute leukemia patients at the time of diagnosis; Measured the expression of 6817 human genes using Affymetrix arrays.")
11
Method: Microarray RNA prepared from cells was hybridized to high-density oligonucleotide Affymetrix microarrays containing probes for 6817 human genes. Samples were subjected to a priori quality control standards regarding the amount of labeled RNA and the quality of the scanned Microarray image.” Eight of 80 leukemia samples were discarded. Samples were rejected if : they yielded less than 15 microgms of biotinylated RNA weak hybridization or if there were visible defects in the array(such as scratches).
.")
12
General method for prediction:
Determine whether there were genes whose expression pattern was strongly correlated with class distinction. Given a collection of known samples, create a class predictor. Test for validity of predictors.

13
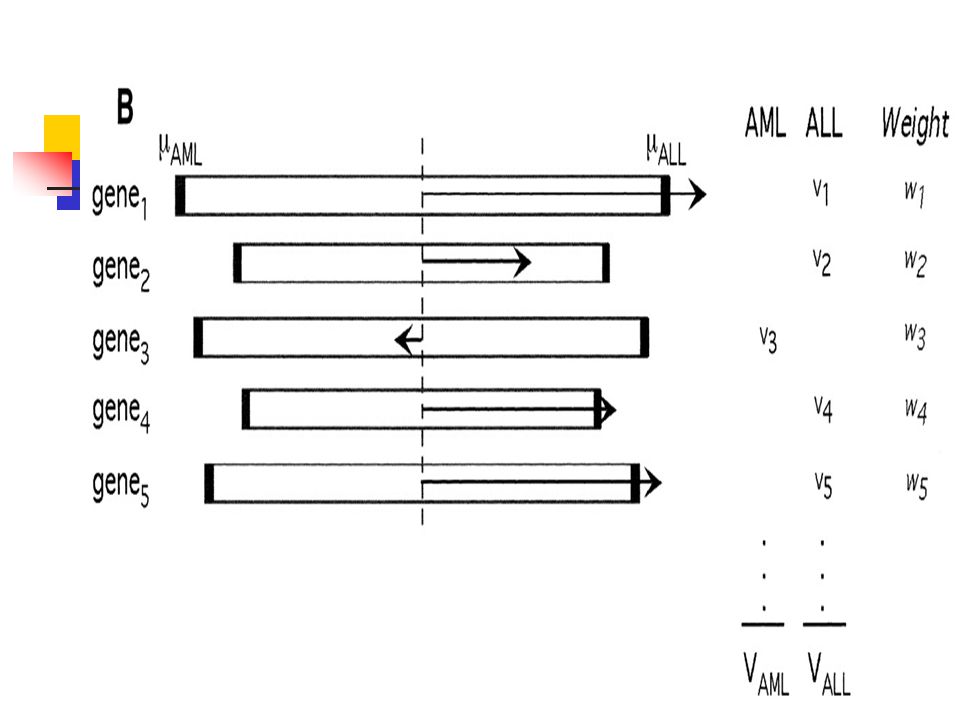
Basic Definitions Each gene is represented by an expression vector
v(g) = ( e1, e2,…….,en) Where ei denotes the expression level of gene g in ith sample . A class distinction is represented by an idealized expression pattern c=(c1,c2,………,cn) where ci is 1 or 0 according to whether the ith sample belongs to class 1 or class 2. “Correlation” between a gene and a class distinction can be measured in a variety of ways.
 = ( e1, e2,…….,en) Where ei denotes the expression level of gene g in ith sample . A class distinction is represented by an idealized expression pattern. c=(c1,c2,………,cn) where ci is 1 or 0 according to whether the ith sample belongs to class 1 or class 2. Correlation between a gene and a class distinction can be measured in a variety of ways.")
15
Correlation A measure of correlation, P(g,c) is used in this paper
P(g,c) = A measure of Correlation measuring the degree to which expression of a given gene in the set of samples correlates with assignment to either class (AML or ALL). [ μ1(g), σ1(g)] and [ μ2(g), σ2(g)] denote the means and SD’s of the log of expression levels of gene g for the samples in class 1 and class2 . P(g,c) = [m1(g) – m2(g) ]/[s1(g) + s2(g)] P(g,c) captures seperation between 2 classes and the variation within individual classes.
 = A measure of Correlation measuring the degree to which expression of a given gene in the set of samples correlates with assignment to either class (AML or ALL). [ μ1(g), σ1(g)] and [ μ2(g), σ2(g)] denote the means and SD’s of the log of expression levels of gene g for the samples in class 1 and class2 . P(g,c) = [m1(g) – m2(g) ]/[s1(g) + s2(g)] P(g,c) captures seperation between 2 classes and the variation within individual classes.")
16
Correlation Cont. Large values of | P(g,c) | indicate a strong correlation between gene expression and the class distinction. P(g,c) being positive or negative corresponds to gene being more highly expressed in class1 or class2.
 being positive or negative corresponds to gene being more highly expressed in class1 or class2.")
17
Method: Neighborhood Analysis

18
Method: Neighborhood Analysis
This method helps establish whether the observed correlation were stronger than would be expected by chance. One defines an "idealized expression pattern" ‘c’ corresponding to a gene that is uniformly high in one class and uniformly low in the other. One tests whether there is an unusually high density of genes "nearby" (or similar to) this idealized pattern, as compared to equivalent random patterns.
 this idealized pattern, as compared to equivalent random patterns.")
19
Each gene is represented by an expression vector, consisting of its expression level in each of the tumor samples. In the fig. The data set is composed of 6 ALLs and 6 AMLs Gene g1 is well correlated with class distinction, whereas gene g2 is poorly correlated.

20
Neighborhood analysis involves counting the number of genes having various levels of correlation with c. The results are compared to the corresponding distribution obtained for random idealized expression patterns c*, obtained by randomly permuting the coordinates of c. Unusually high density of genes indicates that there are many more genes correlated with the pattern than expected by chance.

21
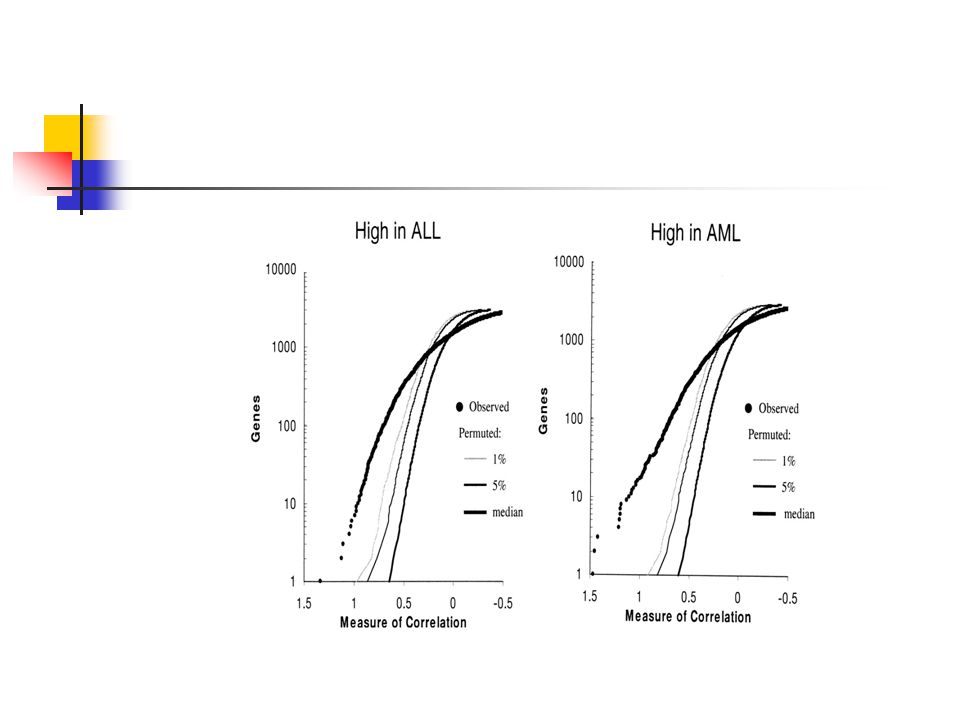
For the 38 acute leukemia samples in the initial data set, neighborhood analysis showed that roughly 1100 genes were more highly correlated with the AML-ALL class distinction than would be expected by chance (Fig. 2). This suggested that classification could indeed be based on expression data.
. This suggested that classification could indeed be based on expression data..")
22
High in ALL: the number of genes with correlation P(g,c) > 0
High in ALL: the number of genes with correlation P(g,c) > 0.30 was 709 for the AML-ALL distinction. Note that P(g,c) = 0.30 is the point where the observed data intersects the 1% significance level.
 > 0.30 was 709 for the. AML-ALL distinction. Note that P(g,c) = 0.30 is the point where the observed data intersects the 1% significance level.")
23
Similarly, in the right panel (higher in AML), 711 genes with P(g,c) > 0.28 were observed.
, 711 genes with P(g,c) > 0.28 were observed.")
24
Choosing a prediction set S
Once the neighborhood analysis graph shows that there are genes significantly correlated with class distinction c , the next step is to choose a set of genes for prediction. The goal is to choose a set S of k genes such that most genes in S are likely to be predictive of the class distinction in future samples.

25
One could simply choose the top k genes by the |P(g,c)|, but there is a possibility that,
for e.g., all genes might be expressed at a high level in class1 and a low level(or not at all) in class 2. They found that the predictors often do better when they include some genes expressed at high levels in each class.
 in class 2. They found that the predictors often do better when they include some genes expressed at high levels in each class.")
26
Choosing S cont’d So, they performed separate neighborhood analysis for positive and negative P(g,c) scores and selected the top k1 genes( highly expressed in class 1) and the bottom k2 genes(highly expressed in class 2).
 and the bottom k2 genes(highly expressed in class 2).")
28
Constraints on selection of k1 and k2.
In order to determine how sensitive our prediction method is to the exact genes used and to choose in choosing k1 and k2 accordingly. There are several constraints :- They wanted to limit the number of genes to those shown to be significantly correlated by the permutation test, perhaps at the 1% level. Furthermore, suppose that there were a single gene whose expression level was perfectly correlated with the class distinction in the available data.

29
We still might like a predictor that includes > 1 gene, in order to provide robustness against noise and to allow to estimate prediction accuracy (described later). Additional genes may be active in different biological pathways or may provide independent estimates of activity along the same pathway; in either case they can add information that can be combined to improve prediction. On the other hand..

30
On the other hand, this argument cannot be extended indefinitely.
If there are 1000 genes that are significantly correlated with a class distinction, it’s unlikely that they all represent different biological mechanisms. Their expression patterns are probably dependent, so that the thousandth gene would be unlikely to add information not already provided by the previous 999, but it would add noise to the system

31
In general, then, there is a tradeoff between
the amount of additional information and robustness gained by adding more genes, and the amount of noise added.

32
Therefore, optimal size of the prediction set is likely to vary somewhat due to the genetics of the class being predicted . (i.e. whether there are many independent classes of genes correlated with the class distinction,or just a single co-regulated pathway; whether many genes or few).
.")
33
Choosing k1 and k2 They evaluated two methods for choosing k1 and k2 :- 1) First method chooses all genes above the 1% significance level in neighborhood analysis, but sets a maximum of 50 genes in each direction to avoid being dominated by noise. 2) The second method tries many different values for |S|,with constraint that k1 and k2 are roughly equal. Preliminary results comparing the two methods indicate that they provide roughly equivalent predictive ability.
 First method chooses all genes above the 1% significance level in neighborhood analysis, but sets a maximum of 50 genes in each direction to avoid being dominated by noise. 2) The second method tries many different values for |S|,with constraint that k1 and k2 are roughly equal. Preliminary results comparing the two methods indicate that they provide roughly equivalent predictive ability.")
34
Questions? How do we use the chosen genes to predict?
How to evaluate the method’s success?

35
After the selection of S , the next step is to try predicting new samples.
They assumed that they have a set of samples called the training set whose correct classifications are already known. And a test set of additional samples whose classes are currently unknown, at least to the algorithm.

36
Prediction by weighted voting
To determine the classification of a new sample in the test set, they used a simple weighted voting scheme. Each gene in S gets to cast its vote for exactly one class. The gene’s vote on a new sample x is weighted by how closely its expression in the training set correlates with c. The vote is the product of this weight and a measure of how informative the gene appears to be for predicting the new sample.

37
Intuitively, we’d expect the gene’s expression in x to look like that of either a typical class-1 sample or a typical class-2 sample in the training set. So, they compared the expression in the new sample to the class means in the training set.

38
They defined a “decision boundary” b (average of the means) halfway between the two class means.
The vote corresponds to the distance between b and the gene’s expression in the new sample. Fig.

40
So, each gene casts a weighted vote
V= weight(g).distance(x,b). They used P(g,c) as weight(g). The weights are defined so that +ve votes->Class1 -ve votes-> class2.
.distance(x,b). They used P(g,c) as weight(g). The weights are defined so that +ve votes->Class1. -ve votes-> class2.")
41
Explanation on Weighted Voting.
Consider a gene represented by gene expression vector g, Let x be the raw expression level of that gene in a new sample whose class we want to predict. Let x~ = log10 x, and let g~= (log10(g1),…, log10(gn)). Let and represent the mean and standard deviation of g~ . Normalized g~norm=((g~1- )/ ,…, (g~n- )/ ) and x~norm = (x~- )/ . (Note that x~norm is normalized by the mean and standard deviation over the training set only.)
,…, log10(gn)). Let and represent the mean and standard deviation of g~ . Normalized g~norm=((g~1- )/ ,…, (g~n- )/ ) and x~norm = (x~- )/ . (Note that x~norm is normalized by the mean and standard deviation over the training set only.)")
42
Class mean for g~norm : 1 = ((I Class1) g~norm )/|class 1| and 2 similarly. b=(1 + 2)/2 V= P(g,c)(x~norm – b). Positive votes are counted as votes for the new sample’s membership in class1, negative votes for class2.
(x~norm – b). Positive votes are counted as votes for the new sample’s membership in class1, negative votes for class2.")
43
To see why this works? Recall, P(g,c) = (1 - 2 )/(1+ 2,).
So, P(g,c) is positive if 1 > 2 and negative if 1 < 2. Suppose, that 1 > 2 , If x looks like a typical sample in class 1, x~norm > b, so x~norm – b > 0 and the weighted vote will be positive. If x looks like a typical sample in class 2, x~norm <b, so x~norm – b < 0 and the weighted vote will be negative. A similar argument holds if 1 < 2 : the signs of (x~norm – b) are reversed but cancel with the negative sign of P(g,c), so the weighted votes are still positive for class 1 and negative for class 2.
 is positive if 1 > 2 and negative if 1 < 2. Suppose, that 1 > 2 , If x looks like a typical sample in class 1, x~norm > b, so x~norm – b > 0 and the weighted vote will be positive. If x looks like a typical sample in class 2, x~norm <b, so x~norm – b < 0 and the weighted vote will be negative. A similar argument holds if 1 < 2 : the signs of (x~norm – b) are reversed but cancel with the negative sign of P(g,c), so the weighted votes are still positive for class 1 and negative for class 2.")
44
The votes for all genes in S combined;
V1 is the sum of all positive votes. V2 is the sum of all negative votes. The winner, and the direction of prediction, is simply the class receiving the larger total vote.

45
Intuitively, If one class receives most of the votes and the other class has only a token representation, it would be reasonable to predict with the majority. However, if the margin of victory is slight, a prediction for the majority class seems somewhat arbitrary and can only be done with a low confidence. They, therefore defined the “ prediction strength” (PS) to measure the margin of victory.
 to measure the margin of victory.")
46
Prediction Strength(PS)
PS= (Vwinner – Vloser)/(Vwinner + Vloser). Since Vwinner is always greater than Vloser, PS varies between 0 and 1. Note: Typical prediction strengths for incorrect predictions tend to be much lower than those for correct predictions.
/(Vwinner + Vloser). Since Vwinner is always greater than Vloser, PS varies between 0 and 1. Note: Typical prediction strengths for incorrect predictions tend to be much lower than those for correct predictions.")
47
The tradeoff between reliability and useful
Thus, the PS measure provides a quantitative way of defining a tradeoff between a “reliable” predictor (one that is always almost correct but sometimes refuses to predict), and a “useful” predictor(one that makes a prediction in every case, but may be incorrect sometimes).
, and a useful predictor(one that makes a prediction in every case, but may be incorrect sometimes).")
48
When to use reliable predictor or useful predictor
If it is essential to make a prediction every time, one can always predict in favor of the winning class, however small the margin of victory.

49
Reliable predictor On the other hand if the cost of an incorrect prediction is high, one can choose a PS threshold below which predictions are not made. For example:-When the predictions are used to diagnose or direct treatment of cancer patients, an incorrect prediction could have potentially devastating results. In this case, they choose a conservatively high PS threshold(0.3) to minimize the chance of making an incorrect diagnosis .(and potentially treating a patient with the wrong chemotherapy regime).
 to minimize the chance of making an incorrect diagnosis .(and potentially treating a patient with the wrong chemotherapy regime).")
50
Evaluating the method The preceding discussion suggests two criteria for evaluating a predictor: The error rate, or percentage of incorrect predictions from the total number of predictions made. The “no-call” rate, or the percentage of samples for which no prediction was made (due to a PS below the threshold).
.")
51
When the number of samples is limited, we evaluate the model by n-way cross validation:
Remove a single sample from the data set, use the remaining n-1 samples as the training set, And test the algorithm’s ability to predict the withheld sample. This process is repeated for each of the n samples in turn, and the error and no call rates are calculated over the entire data set.

52
More on validation When additional samples become available,they use them to test the best model found in the cross validation step. If the initial data set is large enough to divide in two, you may still benefit from a cross-validation step in a number of ways. Try models with different numbers of genes in cross-validation, and pick the best one as the final model to apply to future data.

53
More on validation Could evaluate the tradeoff between error and no-call rate at the cross-validation stage. Plot the cumulative cross-validation rate (w/ a PS threshold of 0) against PS, to determine a reasonable choice for the PS cutoff to use in the test set.
 against PS, to determine a reasonable choice for the PS cutoff to use in the test set.")
54
Test for correlation Build predictor Validate model
Results & GeneCluster Test for correlation Build predictor Validate model

55
Is there correlation? Setup: 38 cell tissue samples, 6817 gene expression levels measured in each, 2 labels (ALL, AML). Create idealized expression vector (c) which correlates exactly with class label. Measure correlation P(c,g) from each gene’s vector to the ideal. Is it significant? Use permutation test to obtain distribution under the null hypothesis. Compare to observed data.
 which correlates exactly with class label. Measure correlation P(c,g) from each gene’s vector to the ideal. Is it significant Use permutation test to obtain distribution under the null hypothesis. Compare to observed data.")
56
Is the correlation significant?

57
Importing data Data from paper available at the authors’ website:
Files all ready for analysis with GeneCluster. Already normalized, though we could reconstruct original. Their method: For each sample s but the first (baseline) { For each gene g with high-quality calls in both { Plot g’s intensity in s and the baseline sample } Fit the best line. Scaling factor for s = 1 / slope of line. Multiply every value in sample s by the scaling factor.
 { For each gene g with high-quality calls in both { Plot g’s intensity in s and the baseline sample. } Fit the best line. Scaling factor for s = 1 / slope of line. Multiply every value in sample s by the scaling factor.")
58
Importing data

59
GeneCluster procedure
Imitating the paper: File --> Open. Separately load both .cls file and .res or .gct file. Dataset --> Preprocess. Transpose data set. Shift global min? Data Analysis --> Marker Selection. Distance function: signal to noise Template: .cls file Class estimate: mean Num neighbors “Run” button to get best correlated genes. Permutation test: 400 perms. “Run”. Examine scores at desired significance threshold. [for reference or emergency only]

60
Data preprocessing

61
Data analysis Explain columns: mean0, std0, mean1, std1, score, class, perm 1%, perm 5%

62
Permutation test Randomly permute class labels (i.e. permute entries of ideal expression vector) n times. Any genes that correlate with the randomized version probably do so by chance. Find the best-correlated k genes each time. Record scores in k bags: list of top-gene scores, list of 2nd-best scores, etc. To find 1% significance level for the best gene, take 1% mark from the list of best genes. To find x% sig level for the kth best gene, take x% value from list of kth best genes. Controls for number of genes ranked (multiple comparison problem) and for correlation between genes.
 n times. Any genes that correlate with the randomized version probably do so by chance. Find the best-correlated k genes each time. Record scores in k bags: list of top-gene scores, list of 2nd-best scores, etc. To find 1% significance level for the best gene, take 1% mark from the list of best genes. To find x% sig level for the kth best gene, take x% value from list of kth best genes. Controls for number of genes ranked (multiple comparison problem) and for correlation between genes.")
63
Correlation confirmed
Paper found 1100 genes more highly correlated with AML-ALL class distinction than by chance. Did not specify significance threshold used. Comparing score vs. 1% and even .005% significance cutoff by eye, I found about 2000 that looked good. I also tried to work with the data on the course website (not normalized though). Instead of showing correlation, the scores were near the median significance level (i.e. chance alone would give scores that high 50% of the time).
. Instead of showing correlation, the scores were near the median significance level (i.e. chance alone would give scores that high 50% of the time).")
64
Test for correlation Build predictor Validate model
Results & GeneCluster Test for correlation Build predictor Validate model

65
Creating class predictor
1100 genes is too many. Restrict to, say, the 50 most correlated genes. Choose half which are high in class 1, half high in class 2. Define parameters (ag, bg) for each gene. weight: ag = P(c,g) vote: bg = [µ1(g) + µ2(g)] / 2 xg = normalized log expression level in new sample Gene contributes vote vg = ag(xg - bg). Predictor outputs class with higher total votes. Prediction strength threshold of 0.3 used.
 for each gene. weight: ag = P(c,g) vote: bg = [µ1(g) + µ2(g)] / 2. xg = normalized log expression level in new sample. Gene contributes vote vg = ag(xg - bg). Predictor outputs class with higher total votes. Prediction strength threshold of 0.3 used.")
66
GeneCluster procedure
Imitating the paper: Dataset --> Build Predictor. 50 features. Two sided = half correlated with each class Class estimate: mean Cross validate: leave one out Algorithm: weighted voting preprocessing: Log10NormFeatures [for reference or emergency only]

67
GeneCluster build predictor window

68
Test for correlation Build predictor Validate model
Results & GeneCluster Test for correlation Build predictor Validate model

69
Cross validation results
Problem: limited data. How to test and improve classifier’s accuracy? Partial solution: cross validation. Divide original data, multiple ways, into mini- training and test sets. Build classifier on training set, check its accuracy on test set. Leave 1 out cross validation. 38 times: Build best predictor using 37 samples. Use it to predict last sample. Classifier made 36 predictions, all of which were correct. Called “uncertain” on remaining 2. (Prediction strength too low.) Now, satisfied with methodology, build final predictor using entire data set.
 Now, satisfied with methodology, build final predictor using entire data set.")
70
Expression level color plot
Top: best correlated 25 genes high in ALL. Bottom: best correlated 25 genes high in AML. Note: no single gene is a perfect predictor. Need a set (at least 10?) for indicator to be robust.
 for indicator to be robust.")
71
Independent test set Independent collection of 34 samples (left from original set of 80). Why didn’t we use it before, when we were complaining about lack of data? Needed this to be truly independent, so a true test of accuracy. Using it earlier (while developing classifier) would bias the classifier to do well on it. Different composition; cell functions not necessarily comparable. 24 specimens bone marrow (like original set), 10 peripheral blood. Derived from both adults (like original set) and children. 31 prepared like originals, 3 processed using very different protocol. The 50-gene predictor was applied. Made calls on 29 of samples. Accuracy: 100%.
 would bias the classifier to do well on it. Different composition; cell functions not necessarily comparable. 24 specimens bone marrow (like original set), 10 peripheral blood. Derived from both adults (like original set) and children. 31 prepared like originals, 3 processed using very different protocol. The 50-gene predictor was applied. Made calls on 29 of samples. Accuracy: 100%.")
72
Test set expression levels
Expression levels of the 50-gene set in the independent test samples.

73
Observations Prediction strengths generally high. Medians 0.77 and 0.73 shown with horizontal lines. Choice to use 50 genes somewhat arbitrary. On this data set, further experiments also achieved 100% accuracy using anywhere from genes. Could experiment with set size on any given data set.

74
Conclusions

75
Future directions Analysis of the 50 informative genes used.
Cell surface proteins (CD11c, CD33, MB-1) already known to distinguish the tumor types. Novel markers (leptin receptor). Known cancer-related genes (Cyclin D3, Op18, . . .). Provide insights for drug design and other cancer study. Classifier method potentially applicable to any measurable distinction among tumors. E.g. predicting clinical outcomes of treatment.
 already known to distinguish the tumor types. Novel markers (leptin receptor). Known cancer-related genes (Cyclin D3, Op18, . . .). Provide insights for drug design and other cancer study. Classifier method potentially applicable to any measurable distinction among tumors. E.g. predicting clinical outcomes of treatment.")
76
Biological significance
Leukemia tumors morphologically and clinically difficult to distinguish, require different treatments. Predictor serves as proof-of-concept that microarray data can be analyzed to learn diagnostic tests.

Similar presentations


>")

















