Download presentation
Presentation is loading. Please wait.

1
High-throughut comparative genomics 24th October 2013 Joe Parker, Queen Mary University London

2
Topics 1.Introduction 2.Background: why phylogenomics? 3.Examples 4.Practice 5.Case study 6.On the horizon 7.Over the horizon

3
Aims Context of phylogenomics: Next- generation sequencing (NGS) Why phylogenomics? Practical analyses Future developments

4
1. Our Research

5
Lab Interests Ecology and evolution of traits Echolocation, sociality NGS data for population genetics and phylogenomics

6
Activities Phylogeny estimation/comparison Molecular correlates of evolution; –site substitutions, dN/dS, composition Simulation Dataset limitations (R-L): Joe Parker; GeorgiaTsagkogeorga; Kalina Davies; Steve Rossiter; Xiuguang Mao; Seb Bailey
: Joe Parker; GeorgiaTsagkogeorga; Kalina Davies; Steve Rossiter; Xiuguang Mao; Seb Bailey")
7
2. Background

8
Next-generation sequencing

9
Why phylogenomics, not - genetics? Causes of discordant signal –Incomplete lineage sorting –Lateral transfer –Recombination –Introgression

10
Quantitative biology Multiple configurations Hyperparameters empirically investigated Determine sensitivity of results

11
Distributions Genome-scale data provides context Identify outliers Genes / taxa / trees Compare values across biological systems

12
Integration with ‘Omics Multiple databases Functional data Bibliographic information

13
3. Example studies

14
Tsakgogeorgia et al. (in press)
")
15
Salichos & Rokas (2013)
")
16
Backström et al. (2013)
")
17
Lindblad-Toh et al. (2011)
")
18
4. Practice

19
Source material Samples Storage Purification Library prep

20
Sequencing Genome –Sanger –Illumina –Pyro /454 –SOLiD –PacBio Transcriptome / RNA-seq –MyBAITS HiSeq / MiSeq IonTorrent

21
Infrastructure Desktop machines Computing clusters Grid systems Cloud-based computation

22
Assembly, Annotation Assembly –To reference (mapping) –De novo Annotation –By homology –De novo SOAPdenovo MAKER Velvet Bowtie / Cufflinks / Tophat Trinity
 –De novo Annotation –By homology –De novo SOAPdenovo MAKER Velvet Bowtie / Cufflinks / Tophat Trinity")
23
Alignment PRANK MUSCLE MAFFT Clustal

24
Phylogeny inference MrBayes RAxML BEAST MP-EST STAR

25
Phylogenetic analysis BEAST HYPHY PAML Pipelines LRT

26
5. Case study

27
Parker et al. (2013) De novo genomes: –four taxa –2,321 protein-coding loci –801,301 codons Published: –18 genomes ~69,000 simulated datasets ~3,500 cluster cores
 De novo genomes: –four taxa –2,321 protein-coding loci –801,301 codons Published: –18 genomes ~69,000 simulated datasets ~3,500 cluster cores.")
28
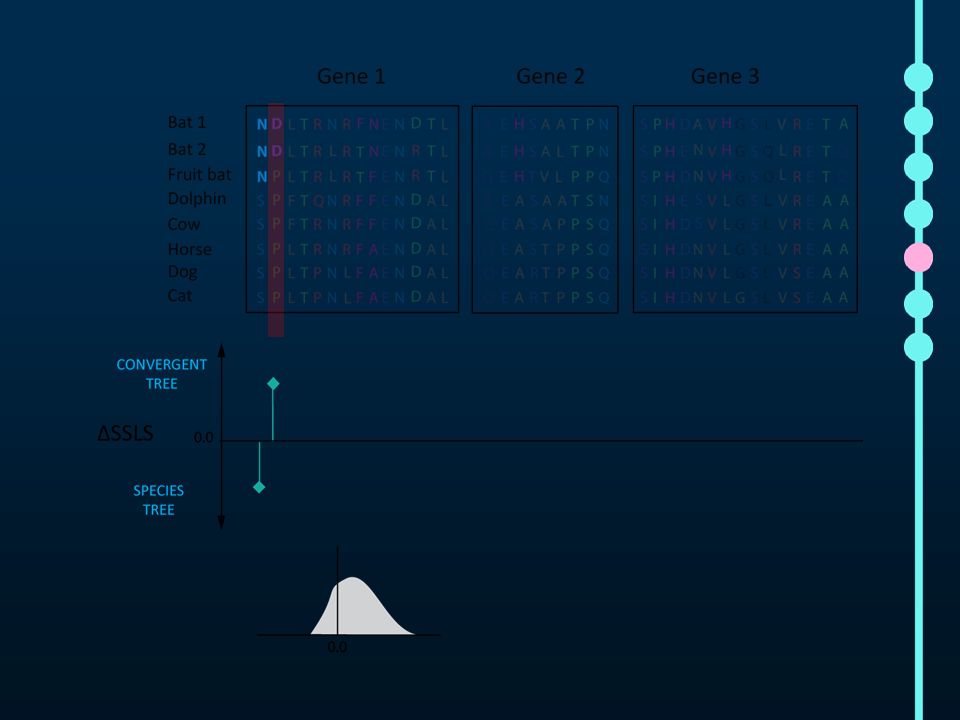
Our pipeline for detecting genome-wide convergence

36
mean = 0.05

37
mean = 0.05mean = -0.01mean = -0.08

38
Development cycle Design Wireframe & specify tests Implement Alignment loadSequences() getSubstitutions() Phylogeny trimTaxa() getMRCA() DataSeries calculateECDF() randomise() Regression getResiduals() predictInterval() Review, refine & refactor
 getSubstitutions() Phylogeny trimTaxa() getMRCA() DataSeries calculateECDF() randomise() Regression getResiduals() predictInterval() Review, refine & refactor")
39
Parker et al. (2013)
")
41
6. On the horizon

42
Environmental metagenomics

43
Models of computation Cloud resources: Unlimited flexibility, finite time Development trade-off –Off-the-shelf –Bespoke Exploratory work –Real time genomic transects? Essential fundamental data missing from nearly every system; –Diversity; structure; substitution rates; dN/dS; recombination; dispersal; lateral transfer

44
Serialisation Process data remotely Freeze-dry objects, download to desktop Implement new methods directly on previously- analysed data

45
7. Over the horizon Real-time phylogenetics Field phylogenetics Alignment-free analyses

46
Conclusions Why phylogenomics? Practice Comparative approach Statistical context

47
Thanks Steve Rossiter 1, James Cotton 2, Elia Stupka 3 & Georgia Tsagkogeorga 1 1 School of Biological and Chemical Sciences, Queen Mary, University of London 2 Wellcome Trust Sanger Institute 3 Center for Translational Genomics and Bioinformatics, San Raffaele Institute, Milan Chris Walker & Dan Traynor Queen Mary GridPP High-throughput Cluster Chaz Mein & Anna Terry Barts and The London Genome Centre Mahesh Pancholi School of Biological and Chemical Sciences BBSRC (UK); Queen Mary, University of London
; Queen Mary, University of London")
48
Resources My email: Joe Parker (Queen Mary University of London): j.d.parker@qmul.ac.ukj.d.parker@qmul.ac.uk Parker, J., Tsagkogeorga, G., Cotton, J.A., Liu, Y., Provero, P., Stupka, E. & Rossiter, S.J. (2013) Genome-wide signatures of convergent evolution in echolocating mammals. Nature 502(7470):228-231 doi:10.1038/nature12511. Tsagkogeorga, G., Parker, J., Stupka, E., Cotton, J.A., & Rossiter, S.J. (2013) Phylogenomic analyses elucidate evolutionary relationships of the bats (Chiroptera) Curr. Biol. in the press. Salichos, L. & Rokas, A. (2013) Inferring ancient divergences requires genes with strong phylogenetic signals. Nature 437:327- 331. doi:10.1038/nature12130 Backström, N., Zhang, Q. & Edwards, S.V. (2013) Evidence from a House Finch (Haemorhous mexicanus) Spleen Transcriptome for Adaptive Evolution and Biased Gene Conversion in Passerine Birds. MBE 30(5):1046-50. doi:10.1093/molbev/mst033 Lindblad-Toh, K., Garber, M., Zuk, O., Lin, M.F., Parker, B.J.,et al. (2011) A high-resolution map of human evolutionary constraint using 29 mammals. Nature 478:476–482 doi:10.1038/nature10530 Degnan, J.H. & Rosenberg, N.A. (2009) Gene tree discordance, phylogenetic inference and the multispecies coalescent. TREE 24:(6)332-340 doi:10.1016/j.tree.2009.01.009 The Tree Of Life: http://phylogenomics.blogspot.co.uk/http://phylogenomics.blogspot.co.uk/ RNA-seq For Everyone: http://rnaseq.uoregon.edu/index.htmlhttp://rnaseq.uoregon.edu/index.html Evo-Phylo: http://www.davelunt.net/evophylo/tag/phylogenomics/http://www.davelunt.net/evophylo/tag/phylogenomics/ OpenHelix: http://blog.openhelix.eu/http://blog.openhelix.eu/ Our blogs: http://evolve.sbcs.qmul.ac.uk/rossiter/ (lab) and http://www.lonelyjoeparker.com/?cat=11 (Joe)http://evolve.sbcs.qmul.ac.uk/rossiter/http://www.lonelyjoeparker.com/?cat=11
 Genome-wide signatures of convergent evolution in echolocating mammals. Nature 502(7470): doi: /nature Tsagkogeorga, G., Parker, J., Stupka, E., Cotton, J.A., & Rossiter, S.J. (2013) Phylogenomic analyses elucidate evolutionary relationships of the bats (Chiroptera) Curr. Biol. in the press. Salichos, L. & Rokas, A. (2013) Inferring ancient divergences requires genes with strong phylogenetic signals. Nature 437: doi: /nature12130 Backström, N., Zhang, Q. & Edwards, S.V. (2013) Evidence from a House Finch (Haemorhous mexicanus) Spleen Transcriptome for Adaptive Evolution and Biased Gene Conversion in Passerine Birds. MBE 30(5): doi: /molbev/mst033 Lindblad-Toh, K., Garber, M., Zuk, O., Lin, M.F., Parker, B.J.,et al. (2011) A high-resolution map of human evolutionary constraint using 29 mammals. Nature 478:476–482 doi: /nature10530 Degnan, J.H. & Rosenberg, N.A. (2009) Gene tree discordance, phylogenetic inference and the multispecies coalescent. TREE 24:(6) doi: /j.tree The Tree Of Life: RNA-seq For Everyone: Evo-Phylo: OpenHelix: Our blogs: (lab) and cat=11 (Joe) cat=11.")
Similar presentations










>")




 Global Ocean Sampling project (GOS) CAMERA CAMERA METAREP.>")





. The Agricultural.>")




 - set of candidate transcripts on which pe read p can be mapped within 1 std. dev. – y(t) -1 if a candidate transcript t is selected,>")