Download presentation
Presentation is loading. Please wait.

1
Drug interactions

2
Drug interactions : another drug, herbal medicine, food, drink or
An interaction is said to occur when the effects of one drug are altered by the co-administration of another drug, herbal medicine, food, drink or other environmental chemical agents

3
Withdrawal Mibefradil terfenadine grepafloxacin Cisapride
Drug interactions are an important cause of prolongation of the QT interval on the electrocardiogram which increases the risk of developing a life-threatening ventricular arrhythmia .

4
MAOIs // Grapefruit juice
The recognition of clinically significant interactions requires knowledge of – pharmacological mechanisms of drug interactions a thorough understanding of high-risk drugs vulnerable patient groups.

5
Susceptible patients The risk of drug interactions increases with the number of drugs used. The rate of ADR in patients taking 6–10 drugs was 7%, rising to 40% in those taking 16–20 drugs, with the exponential rise being largely attributable to drug interactions. In a high-risk group of emergency department patients, the risk of potential adverse drug interaction was 13% in patients taking 2 drugs and 82% in those taking 7 or more drugs (Goldberg et al., 1996). Although polypharmacy is common and often unavoidable, it places certain patient groups at increased risk of drug interactions
. Although polypharmacy is common and often unavoidable, it places certain patient groups at increased risk of drug interactions.")
6
Susceptible patients Patients at particular risk
hepatic or renal disease on long-term therapy for chronic disease (HIV infection, epilepsy, diabetes, patients in intensive care, transplant recipients, patients undergoing complicated surgical procedures Critically ill and elderly patients are Critically ill and elderly patients are at increased risk take more medicines but also because of impaired homeostatic

7
Examples of drugs with high risk of interaction
Concentration-dependent toxicity Digoxin Lithium Aminoglycosides Warfarin Cytotoxic agents Steep dose–response curve Verapamil Sulphonylureas Levodopa

8
Examples of drugs with high risk of interaction
Patient dependent on therapeutic effect Immunosuppressives, e.g., ciclosporin, tacrolimus Glucocorticoids Oral contraceptives Antiepileptics Antiarrhythmics Antipsychotics Antiretrovirals Saturable hepatic metabolism Phenytoin Theophylline

9
Mechanisms of drug interactions
Pharmacokinetic interactions Pharmacokinetic interactions are those that affect the processes by which drugs are - absorbed distributed metabolized or excreted.

10
Changes in gastro-intestinal pH.
Antacids, histamine H2 antagonists and omeprazole can significantly decrease the bioavailability of ketoconazole and itraconazole (fluconazole and voriconazole is not significantly altered) the potential for interaction may be minimised by leaving an interval of 2–3 h between the antacid and the potentially interacting drug. Adsorption, chelation and other complexing mechanisms. tetracyclines and the quinolone antibiotics Most chelation and adsorption interactions can be avoided if an interval of 2–3 h is allowed between doses of the interacting drugs.
 the potential for interaction may be minimised by leaving an interval of 2–3 h between the antacid and the potentially interacting drug. Adsorption, chelation and other complexing mechanisms. tetracyclines and the quinolone antibiotics. Most chelation and adsorption interactions can be avoided if an interval of 2–3 h is allowed between doses of the interacting drugs.")
11
Effects on gastro-intestinal motility.
Anticholinergic agents used in the management of movement disorders have been shown to reduce the bioavailability of levodopa by as much as 50%, possibly as a result of increased metabolism in the intestinal mucosa. Opioids such as diamorphine and pethidine strongly inhibit gastric emptying and greatly reduce the absorption rate of paracetamol, without affecting the extent of absorption. Codeine has no significant effect on paracetamol absorption. Metoclopramide increases gastric emptying and increases the absorption rate of paracetamol, an effect which is used to therapeutic advantage in the treatment of migraine to ensure rapid analgesic effect.

12
Induction or inhibition of drug transport proteins.
The oral bioavailability of some drugs is limited by the action of drug transporter proteins, which eject drugs that have diffused across the gut lining back into the gut. Digoxin is a substrate of P-glycoprotein and drugs that inhibit P-glycoprotein, such as verapamil, may increase digoxin bioavailability with the potential for digoxin toxicity

13
Malabsorption. Drugs such as neomycin may cause a malabsorption syndrome leading to reduced absorption of drugs such as digoxin. Orlistat is a specific long-acting inhibitor of gastric and pancreatic lipases, thereby preventing the hydrolysis of dietary fat to free fatty acids and triglycerides. This can lead to reduced absorption of fat-soluble drugs co-administered with orlistat.

14
Drug metabolism phase I reactions phase II reactions
cytochrome P450 (CYP450) mixed function oxidase system. phase I reactions phase II reactions The CYP450 system comprises 57 isoenzymes CYP1, CYP2, CYP3 and CYP4. responsible for most (about 90%) of the metabolism of commonly used drugs in humans debrisoquine hydroxylase (CYP 2D6) a family number, a subfamily letter and a number for an individual enzyme within the subfamily
 mixed function oxidase system. phase I reactions. phase II reactions. The CYP450 system comprises 57 isoenzymes. CYP1, CYP2, CYP3 and CYP4. responsible for most (about 90%) of the metabolism of commonly used drugs in humans. debrisoquine hydroxylase (CYP 2D6) a family number, a subfamily letter and a number for an individual enzyme within the subfamily.")
15
Polymorphisms Polymorphisms affect metabolism of substrate drugs. The genes that encode specific cytochrome 450 isoenzymes can vary between individuals and, sometimes, ethnic groups. For inter-individual variability in CYP2D6 activity, people may be described according to their ability to metabolize debrisoquine. Poor metabolizers tend to have reduced first-pass metabolism, increased plasma levels and exaggerated pharmacological response to this drug, resulting in postural hypotension. By contrast, ultra-rapid metabolizers may require considerably higher doses for a standard effect. About 5–10% of white Caucasians and up to 2% of Asians and black people are poor metabolizers.

16
CYP3A4 (so similar that they cannot be easily distinguished) CYP3A CYP3A5 most important of all drug-metabolising enzymes - because it is abundant in both the intestinal epithelium and the liver and it has the ability to metabolize a multitude of chemically unrelated drugs from almost every drug class. CYP3A is involved in the metabolism of more than half the therapeutic agents that undergo alteration by oxidation.

17
The effect of a cytochrome 450 isoenzyme on a particular substrate can be altered by interaction with other drugs. Drugs may be themselves substrates for a cytochrome 450 isoenzyme and/or may inhibit or induce the isoenzyme. If a drug is metabolised primarily by a single cytochrome 450 isoenzyme, inhibition or induction of this enzyme would have a major effect on the plasma concentrations of the drug. Example:- if erythromycin (an inhibitor of CYP3A4) is taken by a patient being given carbamazepine (which is extensively metabolised by CYP3A4), this may lead to toxicity due to higher concentrations of carbamazepine.
 is taken by a patient being given carbamazepine (which is extensively metabolised by CYP3A4), this may lead to toxicity due to higher concentrations of carbamazepine.")
18
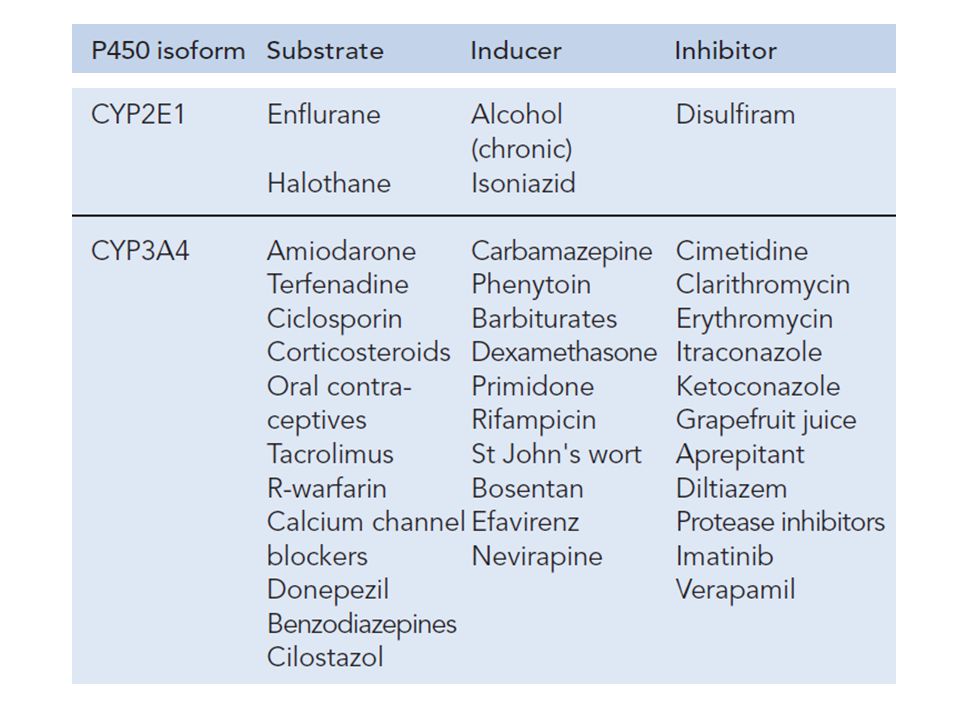
Examples of drug substrates, inducers and inhibitors of the major cytochrome P450 enzymes

19
Examples of drug substrates, inducers and inhibitors of the major cytochrome P450 enzymes

22
Enzyme induction. The most powerful enzyme inducers in clinical use are the antibiotic rifampicin and antiepileptic agents such as barbiturates, phenytoin and carbamazepine. Some enzyme inducers can induce their own metabolism (autoinduction) e.g. barbiturates and carbamazepine. Cigarette smoking, chronic alcohol use and the herbal preparation St John's wort can also induce drug-metabolising enzymes.
 e.g. barbiturates and carbamazepine. Cigarette smoking, chronic alcohol use and the herbal preparation St John s wort can also induce drug-metabolising enzymes.")
23
Since the process of enzyme induction requires new protein synthesis, the effect usually develops over several days or weeks after starting an enzyme-inducing agent and the effect generally persists for a similar period following drug withdrawal. Enzyme-inducing drugs with short half-lives such as rifampicin will induce metabolism more rapidly than inducers with longer half-lives, for example, phenytoin, because they reach steady-state concentrations more rapidly.

24
Enzyme induction usually results in a decreased pharmacological effect of the affected drug.
St John's wort is now known to be a potent inducer of CYP3A When a patient receiving ciclosporin, tacrolimus, HIV-protease inhibitors, irinotecan or imatinib takes St John's wort, there is a risk of therapeutic failure with the affected drug. If the affected drug has active metabolites, this may lead to an increased pharmacological effect.

25
Examples of interactions due to enzyme induction

26
Enzyme inhibition. Enzyme inhibition is responsible for many clinically significant interactions. A strong inhibitor is one that can cause ≥5-fold increase in the plasma area-under-the-curve (AUC) value or more than 80% decrease in clearance of CYP3A substrates. A moderate inhibitor is one that can cause ≥2- but <5-fold increase in the AUC value or 50–80% decrease in clearance of sensitive CYP3A substrates A weak inhibitor is one that can cause ≥1.25- but <2-fold increase in the AUC values or 20–50% decrease in clearance of sensitive CYP3A substrates
 value or more than 80% decrease in clearance of CYP3A substrates. A moderate inhibitor is one that can cause ≥2- but <5-fold increase in the AUC value or 50–80% decrease in clearance of sensitive CYP3A substrates. A weak inhibitor is one that can cause ≥1.25- but <2-fold increase in the AUC values or 20–50% decrease in clearance of sensitive CYP3A substrates.")
27
Examples of interactions due to enzyme inhibition

28
Elimination interactions
Most drugs are excreted in either the bile or urine. Changes in urinary pH. Urine alkalinisation or acidification has been used as a means of increasing drug elimination in poisoning with salicylates and amphetamines, respectively. Changes in active renal tubule excretion. Drugs that use the same active transport system in the kidney tubules can compete with one another for excretion Probenecid may be given to increase the plasma concentration of penicillins by delaying renal excretion. Increased methotrexate toxicity, sometimes life-threatening, has been seen in some patients concurrently treated with salicylates and some other non-steroidal anti-inflammatory drugs (NSAIDs).
.")
29
Elimination interactions
Changes in renal blood flow. Blood flow through the kidney is partially controlled by the production of renal vasodilatory prostaglandins. If the synthesis of prostaglandins is inhibited by drugs such as indometacin, the renal excretion of lithium is reduced with a subsequent rise in plasma levels. Biliary excretion and the enterohepatic shunt. Interaction between broad-spectrum antibiotics and oral contraceptives. Drug transporter proteins.

30
Elimination interactions
P-glycoprotein (P-gp) Drug transporter proteins. P-glycoprotein acts as an efflux pump, exporting s ubstances into urine, bile and the intestinal lumen. Examples of inhibitors and inducers of P-glycoprotein
 Drug transporter proteins. P-glycoprotein acts as an efflux pump, exporting s ubstances into urine, bile and the intestinal lumen. Examples of inhibitors and inducers of. P-glycoprotein.")
31
Pharmacodynamic interactions
It is to be expected that a drug with an agonist action at a particular receptor type will interact with antagonists at that receptor. naloxone flumazenil. α-Adrenergic agonists such as metaraminol may be used in the management of priapism induced by α-adrenergic antagonists such as phentolamine.

32
Additive or synergistic interactions
Examples of additive or synergistic interactions

33
Drug or neurotransmitter uptake interactions
MAOIs Drug–food interactions Grapefruit juice inhibits intestinal CYP3A4 Drug–herb interactions A number of herbal products have anti-platelet and anticoagulant properties and may increase the risk of bleeding when used with aspirin or warfarin.

34
Mrs C is a 62-year-old woman with a history of hypertension, atrial fibrillation and type 2 diabetes. She is a non-smoker and obese. Her current medication comprises flecainide 100 mg twice a day, aspirin 75 mg daily, simvastatin 40 mg and diltiazem 180 mg daily. Mrs C is suffering from a respiratory tract infection and her primary care doctor has prescribed a 5-day course of clarithromycin. Questions 1. Are there likely to be any clinically significant drug interactions? 2. What advice do you give?

35
There is potential for interaction between simvastatin and diltiazem and between simvastatin and clarithromycin. Some statins, particularly simvastatin and atorvastatin, are metabolised by cytochrome P450 (CYP3A4) and co-administration of potent inhibitors of this enzyme may particularly increase plasma levels of these statins and so increase the risk of dose-related side effects, including rhabdomyolysis. Clarithromycin is a potent inhibitor of CYP3A4 and diltiazem is a less potent inhibitor.

36
2. Current advice is that diltiazem and simvastatin may be given together provided the simvastatin dose does not exceed 40 mg daily, so it is reasonable for this therapy to be continued. However, clarithromycin should not be given together with simvastatin. Myopathy and rhabdomyolysis have been reported in patients taking the combination. Mrs C should be advised not to take her simvastatin while she is taking clarithromycin and to start taking it again after she has completed the course of antibiotic.

Similar presentations

























 with age The elderly are presumed to be - because of.>")



