Download presentation
Presentation is loading. Please wait.

1
The Organic Chemistry of Drug Design and Drug Action
Chapter 2 Lead Discovery and Lead Modification

2
Lead Discovery Approaches
1. Random screening - only approach before 1935; screen every compound you have; still a useful approach; streptomycin and tetracyclines identified in this way 2. Nonrandom (or Targeted or Focused) screening - only screen compounds related to active compounds 3. Drug metabolism studies - metabolites produced are screened for the same or other activities 4. Clinical observations - new activities found in clinical trials; Dramamine tested as antihistamine (allergy) - found to relieve motion sickness; Viagra tested as antihypertensive - found to treat erectile dysfunction
 screening - only screen compounds related to active compounds. 3. Drug metabolism studies - metabolites produced are screened for the same or other activities. 4. Clinical observations - new activities found in clinical trials; Dramamine tested as antihistamine (allergy) - found to relieve motion sickness; Viagra tested as antihypertensive - found to treat erectile dysfunction.")
3
Lead Discovery Approaches (cont’d)
5. Rational approaches - identify causes for disease states: imbalance of chemicals in the body invasion of foreign organisms aberrant cell growth Identify biological systems involved in disease states; use natural receptor ligand or enzyme substrate as the lead; a known drug also can be used as a lead

4
Rational Drug Design Chemical imbalances - antagonism or agonism of a receptor; enzyme inhibition Foreign organism and aberrant cell growth - enzyme inhibition; DNA interaction

5
Dopamine as a lead compound
Figure 2.1 Depiction of dopamine (DA) in its role as a neurotransmitter. DA is released by a neuron prior to interacting with dopamine receptors (D1–D5) on the surface of another nearby neuron. Also shown is the dopamine transporter, which terminates the action of dopamine by transporting the released neurotransmitter from the synaptic cleft back into the presynaptic neuron. Reprinted by permission from Macmillan Publishers Ltd: Nature Reviews Drug Discovery (Kreek, M. J.; LaForge, K. S.; Butelman, E. Pharmacotherapy of addictions. Nat. Rev. Drug Discov. 2002, 1, 710–726) Copyright 2002.
 in its role as a neurotransmitter. DA is released by a neuron prior to interacting with dopamine receptors (D1–D5) on the surface of another nearby neuron. Also shown is the dopamine transporter, which terminates the action of dopamine by transporting the released neurotransmitter from the synaptic cleft back into the presynaptic neuron. Reprinted by permission from Macmillan Publishers Ltd: Nature Reviews Drug Discovery (Kreek, M. J.; LaForge, K. S.; Butelman, E. Pharmacotherapy of addictions. Nat. Rev. Drug Discov. 2002, 1, 710–726) Copyright")
6
Leads based on neurotransmitters

7
Example of Rational Drug Design
Serotonin, a mediator of inflammation, was used as the lead for the anti-inflammatory drug indomethacin. indomethacin N C H 3 O l N H 2 O

8
Steroid hormones as lead compounds

9
Peptide hormones as lead compounds

10
Transporter ligands as lead compounds

11
Enzyme substrates as lead compounds
Physostigmine

12
Kinases as targets—ATP is the lead compound
Scheme 2.1 Reaction catalyzed by the kinase class of enzymes. Kinases catalyze the transfer of the terminal phosphate group of ATP or related molecules acceptor to the group of a substrate, in this case, an alcohol (ROH).
.")
13
Other and repurposed ligands as lead compounds
AIDS -> antihistamine Antihypertensive -> Parkinson’s Antidepressant -> back pain

14
Leads from screening

15
Sources of compounds for screening
Natural products Collections of compounds High-throughput synthesis of a library of compounds Solid-phase synthesis Solution-phase synthesis

16
Natural product leads

17
Natural products seem to provide greater structural diversity than combinatorial chemistry.
Natural products that are biologically active in assays generally have drug-like properties. Construction of chemical libraries based on natural product hits is a good compromise.

18
Solid-Phase Synthesis of a Natural Product-like Combinatorial Library
Library based on the 2,2-dimethylbenzopyran scaffold, a privileged structure.

19
Solid-phase synthesis is used to make libraries of compounds
Scheme 2.2 Solid-phase synthesis of a library of 7-hydroxybenzodiazepines

20
Solid phase synthesis of a library
Scheme 2.3 Solid-phase synthesis of a library of 4-alkoxyproline derivatives

21
Combinatorial Chemistry
Synthesis or biosynthesis of chemical libraries of molecules for lead discovery or lead modification Libraries prepared in a systematic and repetitive way by covalent assembly of building blocks to give diversity within a common scaffold. Synthesis carried out on a solid support (polymer resin). Isolation and purification of product of each step done by filtration and washing. Everything not attached to polymer is washed away, which allows use of excess reagents.
. Isolation and purification of product of each step done by filtration and washing. Everything not attached to polymer is washed away, which allows use of excess reagents.")
22
Alternatives: Carry out reactions in solution with excess reagents, which are scavenged with a polymer-bound scavenger. Filtration removes excess reagents bound to polymer. Or, use polymer-bound reagents. Main differences among various methods of combinatorial library synthesis: 1. Solid support used 2. Methods for assembling the building blocks 3. The state (immobilized or in solution) in which libraries are screened 4. Manner in which structures of active compounds are determined (encoding methods)
 in which libraries are screened. 4. Manner in which structures of active compounds are determined (encoding methods)")
23
If the # of building blocks varies in each step, then N = bcd...
# synthetic steps N = bx (Eq. 2.1) # compounds in library # building blocks in each step If the # of building blocks varies in each step, then N = bcd... For example, consider a 4-step synthesis. If 25 different reactants (building blocks) are used in the first two steps, 20 in step three, and 15 in step four, there would be 25 25 20 15 = 187,500 different compounds (from only 85 different building blocks).
 # compounds in library. # building blocks in each step. If the # of building blocks varies in each step, then N = bcd... For example, consider a 4-step synthesis. If 25 different reactants (building blocks) are used in the first two steps, 20 in step three, and 15 in step four, there would be 25 25 20 15 = 187,500 different compounds (from only 85 different building blocks).")
24
Peptide Libraries Split synthesis
Also called mix and split or split and pool method Most common lead discovery approach for large libraries ( compounds), assayed as library mixtures. Produces a collection of polymer beads, each containing one library member ( pmol) A combinatorial library of all pentapeptides comprised of the 20 common amino acids: 205 = 3.2 106 peptides. For example, prepare a homogenous mixture of all tripeptides of His, Val, and Ser (33 = 27)
, assayed as library mixtures. Produces a collection of polymer beads, each containing one library member ( pmol) A combinatorial library of all pentapeptides comprised of the 20 common amino acids: 205 = 3.2 106 peptides. For example, prepare a homogenous mixture of all tripeptides of His, Val, and Ser (33 = 27)")
25
Split Synthesis of All Tripeptides Containing His, Val, and Ser

26
Split Synthesis of Most Active Hexapeptide for a Opioid Receptor
A combinatorial library is prepared of pentapeptides (comprised of the 20 main amino acids) bound to the MBHA polystyrene resin (makes peptide amides when cleaved off of resin) Separate into 20 tubes, then add a different N-acetylamino acid to each tube Assay the 20 combinatorial library mixtures (either still attached to bead or after cleavage) Set the N-terminal amino acid of most potent library constant
 bound to the MBHA polystyrene resin (makes peptide amides when cleaved off of resin) Separate into 20 tubes, then add a different N-acetylamino acid to each tube. Assay the 20 combinatorial library mixtures (either still attached to bead or after cleavage) Set the N-terminal amino acid of most potent library constant.")
27
If the most potent was the library with Ac-Arg at N-terminus,
Set all to Arg Vary 2nd amino acid Next 4 amino acids are a combinatorial library Determine best residue at 2nd position, then repeat process for 3rd position. Continue until you get most potent N-acetyl hexapeptide amide (in this case, AcArg-Phe-Met-Trp-Met-Thr-NH2).
.")
28
Problems with Split Synthesis Approach
Most potent analog may be less potent than what was observed in a previous iteration Peptide-peptide interactions may have been responsible for previous potency (some peptides removed in next step) Active compound may be a complex of more than one peptide (some peptides removed in next step) Conformation may be different with fewer peptides present
 Active compound may be a complex of more than one peptide (some peptides removed in next step) Conformation may be different with fewer peptides present.")
29
Nonpeptide Libraries Problems with Assaying Multiple Compounds Together
Many false negatives (active compound that does not produce a hit) and false positives (inactive compound that does produce a hit) observed. Therefore, single entity screens are used. Parallel synthesis - solid-phase reactions carried out to make individual compounds rapidly (and robotically) The library ( compounds) is made in parallel without combining tubes so each support has one compound attached. The goal is to get as much molecular diversity with as much functionality as possible.
 and false positives (inactive compound that does produce a hit) observed. Therefore, single entity screens are used. Parallel synthesis - solid-phase reactions carried out to make individual compounds rapidly (and robotically) The library ( compounds) is made in parallel without combining tubes so each support has one compound attached. The goal is to get as much molecular diversity with as much functionality as possible.")
30
Solution phase synthesis of a library
Scheme 2.4 Solution-phase synthesis of a library of furanose derivatives

31
Libraries can be made in microtiter plates
Figure 2.2 (A) Schematic of a typical 96-well microtiter plate. (Reprinted with permission from Custom Biogenic Systems ( (B) Picture of a 96-well microtiter plate taken by Jeffrey M. Vinocur, 4/21/06, published on Wikipedia Commons (
 Schematic of a typical 96-well microtiter plate. (Reprinted with permission from Custom Biogenic Systems ( (B) Picture of a 96-well microtiter plate taken by Jeffrey M. Vinocur, 4/21/06, published on Wikipedia Commons (")
32
Libraries can be made in microtiter plates
Figure 2.4 Image of 96-well filter plates. Reprinted with permission from Norgen Biotek Corp.

33
Polymer-bound scavenger reagents can be used in synthesis
Figure 2.3 Product of polymer-bound N-methylglucosamine with borate anion

34
Another solid-phase scavenger
Scheme 2.5 Use of a solid-phase scavenger in solution-phase synthesis. In this example, a polymer-bound isocyanate is used to scavenge excess primary or secondary amine from a solution by forming the corresponding polymer-bound urea.

35
An antitumor drug discovered by combinatorial synthesis and high-throughput screening

36
Advantages and problems in high throughput synthesis
Solid-phase synthesis can make large numbers of compounds, but purification is problematic and quantities are small. Solution-phase synthesis makes smaller numbers of compounds, but larger quantities and easier purification.

37
Properties that Influence Oral Bioavailability Lipinski’s Rule of 5
The “rule of 5” states that poor oral absorption and/or distribution are more likely when: The molecular weight is > 500 The log P is > 5 There are more than 5 H-bond donors (expressed as the sum of OH and NH groups) There are more than 10 H-bond acceptors (expressed as the sum of N and O atoms) It is highly likely (>90%) that compounds with two or more of these characteristics will have poor absorption properties. Antibiotics, antifungals, vitamins, and cardiac glycosides are the exception because they often have active transporters to carry them across membranes.
 There are more than 10 H-bond acceptors (expressed as the sum of N and O atoms) It is highly likely (>90%) that compounds with two or more of these characteristics will have poor absorption properties. Antibiotics, antifungals, vitamins, and cardiac glycosides are the exception because they often have active transporters to carry them across membranes.")
38
Alternative to Lipinski’s Rule of Five Veber and co-workers
Poor oral bioavailability found as a result of increased molecular flexibility (independent of molecular weight), as measured by: greater than 10 rotatable bonds high polar surface area (> 140 Å2) total hydrogen bond count (> a total of 12 donors and acceptors) Both the number of rotatable bonds and hydrogen bond count tend to increase with molecular weight, which may explain Lipinski’s first rule. Reduced polar surface area was found to correlate better with an increased permeation rate than did lipophilicity.
, as measured by: greater than 10 rotatable bonds. high polar surface area (> 140 Å2) total hydrogen bond count (> a total of 12 donors and acceptors) Both the number of rotatable bonds and hydrogen bond count tend to increase with molecular weight, which may explain Lipinski’s first rule. Reduced polar surface area was found to correlate better with an increased permeation rate than did lipophilicity.")
39
Privileged Structures and Drug-like Molecules
Certain scaffolds are capable of binding to multiple receptor targets Appropriate structural modifications can change the activity N O Y R H C ' Privileged structure benzodiazepines (a) Evans, B. E.; Rittle, K. E.; Bock, M. G.; DiPardo, R. M.; Freidinger, R. M.; Whitter, W. L.; Lundell, G. F.; Veber, D. F.; Anderson, P. S.; Chang, R. S. L.; Lotti, V. J.; Cerino, D. J.; Chen, T. B.; Kling, P. J.; Kunkel, K. A.; Springer, J. P.; Hirshfield, J. J. Med. Chem. 1988, 31, (b) Ariëns, E. J.; Beld, A. J.; Rodrigues de Miranda, J. F.; Simonis, A. M. in The Receptors: A Comprehensive Treatise; O'Brien, R. D., Ed.; Plenum Press: New York, 1979, p. 33. (b) Ariëns, E. J. Med. Res. Rev. 1987, 7, 367. (c) Patchett, A. A.; Nargund, R. P. Annu. Rep. Med. Chem. 2000, 35, 289. (d) Hajduk, P. J.; Bures, M.; Praestgaard, J.; Fesik, S. W. J. Med. Chem. 2000, 43, (e) Fecik, R. A.; Frank, K. E.; Gentry, E. J.; Menon, S. R.; Mitscher, L. A.; Telikepalli, Med. Res. Rev. 1998, 18, 149. (f) Horton, D. A.; Bourne, G. T.; Smythe, M. L. J. Computer-Aided Mol. Des. 2002, 16, 415. (g) Klabunde, T.; Hessler, B. Chembiochem. 2002, 3, 928. (h) Matter, H. Baringhaus, K.H.; Naumann, T.; Klabunde, T.; Pirard, B. Comb. Chem. High Throughput Screen. 2001, 4, 453. (i) Horton, D. A.; Bourne, G. T.; Smythe, M. L. Chem. Rev. 2003, 103, 893.
 Evans, B. E.; Rittle, K. E.; Bock, M. G.; DiPardo, R. M.; Freidinger, R. M.; Whitter, W. L.; Lundell, G. F.; Veber, D. F.; Anderson, P. S.; Chang, R. S. L.; Lotti, V. J.; Cerino, D. J.; Chen, T. B.; Kling, P. J.; Kunkel, K. A.; Springer, J. P.; Hirshfield, J. J. Med. Chem. 1988, 31, (b) Ariëns, E. J.; Beld, A. J.; Rodrigues de Miranda, J. F.; Simonis, A. M. in The Receptors: A Comprehensive Treatise; O Brien, R. D., Ed.; Plenum Press: New York, 1979, p. 33. (b) Ariëns, E. J. Med. Res. Rev. 1987, 7, 367. (c) Patchett, A. A.; Nargund, R. P. Annu. Rep. Med. Chem. 2000, 35, 289. (d) Hajduk, P. J.; Bures, M.; Praestgaard, J.; Fesik, S. W. J. Med. Chem. 2000, 43, (e) Fecik, R. A.; Frank, K. E.; Gentry, E. J.; Menon, S. R.; Mitscher, L. A.; Telikepalli, Med. Res. Rev. 1998, 18, 149. (f) Horton, D. A.; Bourne, G. T.; Smythe, M. L. J. Computer-Aided Mol. Des. 2002, 16, 415. (g) Klabunde, T.; Hessler, B. Chembiochem. 2002, 3, 928. (h) Matter, H. Baringhaus, K.H.; Naumann, T.; Klabunde, T.; Pirard, B. Comb. Chem. High Throughput Screen. 2001, 4, 453. (i) Horton, D. A.; Bourne, G. T.; Smythe, M. L. Chem. Rev. 2003, 103, 893.")
40
Drug-likeness Only 32 scaffolds describe half of all known drugs.1
Average number of side chains per molecule is 4. If carbonyl is ignored, then 73% of the side chains in drugs are from top 20 most common side chains.2 Drug-likeness may be an inherent property of some molecules.3 Try these first. 1 Bemis, G. W.; Murcko, M. A. J. Med. Chem. 1996, 39, 2887. 2 Bemis, G. W.; Murcko, M. A. J. Med. Chem. 1999, 42, 5095. 3 Ajay; Walters, W. P.; Murcko, M. A. J. Med. Chem. 1998, 41, 3314.

41
Privileged structures
Figure 2.5 Pairs of compounds containing a privileged structure (indole, dihydropyridine, or benzimidazole) and binding to diverse target classes
 and binding to diverse target classes.")
42
Some groups should be avoided because of possible toxicity

43
Sucralose is an alkyl halide, but was approved as a non-nutritive sweetener

44
Many nondrug-like molecules show up as active in screens - false positives.
Appear to bind to numerous receptors, but not at the site of the natural ligand (called promiscuous drugs) May be the result of aggregate formation
 May be the result of aggregate formation.")
45
Virtual screening can accelerate lead identification
Scheme 2.6 The process of virtual screening to identify compounds that conform to a hypothesis specifying properties (that are discernible from a compound’s structure) that are required for activity
 that are required for activity.")
46
Virtual screening databases are available
Tripos LeadQuest Maybridge Accelrys ACD ZINC-a free database

47
Virtual screening process
Identify substructure linked to desired activity Search database for molecules with active substructure 2D search just matches the structure 3D search can match molecular shape

48
Substructure search problems
Figure 2.6 Hypothetical example illustrating that substructure search (e.g., using pyridine as the search query) may not retrieve the most structurally similar compounds in a compound collection
 may not retrieve the most structurally similar compounds in a compound collection.")
49
How to quantify similarity?
T = N11/n-N00 = 7/18-4 = 0.50

50
Computer-Based Methods of QSAR
3D-QSAR correlates diverse molecular structures and their biological function at a particular target. General approach assay a set of molecules align molecules according to some predetermined orientation rules calculate a set of spatially dependent parameters for each molecule determined in the receptor space derive a function relating each molecule’s spatial parameters to their biological property establish self-consistency and predictability of the function

51
Comparative Molecular Field Analysis (CoMFA)
Molecule-receptor interactions are represented by steric and electrostatic fields exerted by each molecule A series of active compounds are identified and 3-D structural models constructed These structures are superimposed on one another and placed within a regular 3-D grid A probe atom placed at lattice points on grid where it is used to calculate steric and electrostatic potentials between itself and each superimposed structure A 3-D contour map is constructed to identify where lower or higher steric or electrostatic interactions increase binding

52
A pharmacophore based on known ligands
Figure 2.7 Example of a computer-generated pharmacophore model. From Laurini et al Bioorg. Med. Chem. Lett (Ref. 111)
")
53
Molecular Graphics Use of computers to display and manipulate molecular structures based on Fischer lock-and-key hypothesis. synonymous names for the method Structure-based drug design Computer-assisted drug design Computer-assisted molecular design Commercial software, such as Sybyl (Tripos), Insight II (Molecular Simulations), and Gold (Cambridge Crystallographic Data Center), are used. Need a high-resolution (crystal or solution) structure of a receptor with a ligand bound. The ligand can be removed graphically to expose the receptor binding site. Other molecules are docked into the binding site.
, Insight II (Molecular Simulations), and Gold (Cambridge Crystallographic Data Center), are used. Need a high-resolution (crystal or solution) structure of a receptor with a ligand bound. The ligand can be removed graphically to expose the receptor binding site. Other molecules are docked into the binding site.")
54
Haldol, a lead for HIV-1 protease inhibitors from docking

55
From “hit” to lead Screening results in hits
In HTS, need to identify and confirm structure of hits In virtual screening, need to perform assays to confirm activity A series of analogs may be prepared Ligand efficiency = ΔG/N

56
Fragment-based identification of leads

57
Comparison of ligand and fragments
Ki 1 μM 40 mM 19 mM 10 mM But structural studies show that the fragments do not bind in the same site

58
SAR by NMR NMR approach to identify and optimize high-affinity ligands bound to proteins
Figure 2.8 SAR by NMR methodology Screen ~10 compounds at a time. Look for a 15N-chemical shift in the protein NMR indicating compound bound at that amino acid in the protein. Once lead is identified, make a focused library of analogs to optimize lead. Screen a 2nd library to find one that binds to a site near to where the first compound bound. Optimize the 2nd lead. Link the two compounds together - binding affinity is dramatically increased.

59
Example of SAR by NMR Inhibition of stromelysin, a matrix metalloprotease (degrades extracellular matrix components in tissue remodeling and in arthritis, osteoporosis, and cancer) Site 1 Site 2 Linker Kd = 17 mM Kd = 20 M Kd = 17 nM Took 6 months to identify Prior to this, no leads had been identified.
 Site 1. Site 2. Linker. Kd = 17 mM. Kd = 20 M. Kd = 17 nM. Took 6 months to identify Prior to this, no leads had been identified.")
60
A clinical candidate from SAR by NMR

61
SAR by NMR What’s Really Involved?
To see a 15N chemical shift in NMR spectrum: 1. Overexpress protein in microorganism grown on 15NH4Cl as sole N source to incorporate 15N in all amino acids 2. Purify protein (need > 200 mg per spectrum) 3. Determine 3D structure of protein by various NMR techniques (mass < 40 kDa) If structure can be determined, SAR by NMR can screen 1000 compounds a day.
 3. Determine 3D structure of protein by various NMR techniques (mass < 40 kDa) If structure can be determined, SAR by NMR can screen 1000 compounds a day.")
62
Generating a lead from fragments
Figure 2.9 Ellman combinatorial methodology for lead generation with an unknown or impure protein

63
SAR by MS High-throughput mass spectrometry-based screen
A set of diverse compounds screened by mass spectrometry to identify those that bind to a receptor Competition experiments determine compounds bound to different sites (ternary complex if bound to different sites; binary complex if at same site) For molecules bound to different sites, vary substituent size to determine which bind at adjacent sites Attach the two molecules with a linker
 For molecules bound to different sites, vary substituent size to determine which bind at adjacent sites. Attach the two molecules with a linker.")
64
Substrate activity screening (SAS)
Figure 2.10 Steps of SAS for identification of protease inhibitors

65
Linking fragments Figure 2.11 Three approaches to linking fragments: (A) fragment evolution, (B) fragment linking, and (C) fragment self-assembly. Reprinted with permission from Macmillan Publishers Ltd: Nature Reviews Drug Discovery (Reese, D. C.; Congreve, M.; Murray, C. W.; Carr, R. Fragment-based lead discovery. Nat. Rev. Drug Discov. 2004, 3, 660–672) Copyright 2004.
 fragment evolution, (B) fragment linking, and (C) fragment self-assembly. Reprinted with permission from Macmillan Publishers Ltd: Nature Reviews Drug Discovery (Reese, D. C.; Congreve, M.; Murray, C. W.; Carr, R. Fragment-based lead discovery. Nat. Rev. Drug Discov. 2004, 3, 660–672) Copyright")
66
Fragment evolution Figure 2.12 Example of fragment-based lead discovery incorporating the fragment evolution approach followed by lead modification to an optimized compound. Ligand efficiencies help guide the overall process.

67
Fragment self-assembly—click chemistry
Figure 2.13 Example of fragment-based lead discovery incorporating the fragment self-assembly approach: click chemistry

68
Leads for sleeping sickness
Figure 2.14 (A) Structure of 2.51 complexed with 6-phosphogluconate dehydrogenase (6PGDH) determined by X-ray crystallography. (B) Structure of 2.53 complexed with 6PGDH predicted by computational docking. From Ruda, et al. Bioorg. Med. Chem. 2010, 18, 5056–5062.
 Structure of 2.51 complexed with 6-phosphogluconate dehydrogenase (6PGDH) determined by X-ray crystallography. (B) Structure of 2.53 complexed with 6PGDH predicted by computational docking. From Ruda, et al. Bioorg. Med. Chem. 2010, 18, 5056–5062.")
69
Fragment-based development of a urokinase-type plasminogen activator inhibitor
Figure 2.15 Progression from mexiletin (2.55, identified by fragment-based screening) to a potent orally bioavailable uPA
 to a potent orally bioavailable uPA.")
70
Lead Optimization Pharmacodynamics receptor interactions - structure of lead is similar to that of the natural receptor ligand or enzyme substrate Pharmacokinetics ADME - Absorption, Distribution, Metabolism, Excretion; depends on water solubility and lipid solubility Toxicity

71
Identification of the Active Part of the Lead
First consider pharmacodynamics Pharmacophore - the relevant groups on the compound that interact with the receptor and produce activity Auxophore - the rest of the molecule

72
Some of the auxophoric groups interfere with binding of pharmacophoric groups and must be excised.
Some of the auxophoric groups may neither bind to the receptor nor prevent the pharmacophoric groups from binding - these can be modified without effect on potency. Modification of these auxophoric groups may be used to correct ADME problems.

73
One approach to determine which are pharmacophoric atoms, which are auxophoric atoms hindering binding, and which are auxophoric atoms that can be modified is to cut away pieces of the lead and measure the effect on potency.

74
Example of Pharmacophore and Auxophore Identification
Assume the well-known addictive analgesics below are the lead compounds. The darkened part is known to be the pharmacophore (you would not know that in a real-life example). All bind to the opioid receptor What happens if we excise the bridging O (shown above not in the pharmacophore)?
. All bind to the opioid receptor. What happens if we excise the bridging O (shown above not in the pharmacophore)")
75
Maybe additional conformational flexibility
3-4 times more potent than morphine Note: The structure is drawn to mimic that of morphine; it is not the lowest energy conformer. Maybe additional conformational flexibility allows the molecule to bind more effectively.

76
Next, remove half of the cyclohexene ring (also not in the pharmacophore).
Less potent than morphine, but much lower addictive properties. about the same potency as morphine

77
Removal of more rings Demerol Potency similar to morphine
10-12% the potency of morphine

78
If every cut gives lower potency, either most of the molecule is in the pharmacophore or the cut causes a conformational change away from the bioactive conformation. Add groups to increase the pharmacophore. 3200 times more potent than morphine

79
Fentanyl and derivatives are the most potent opiates known
Fentanyl and derivatives are the most potent opiates known. Is the ester group in carfentanil part of the pharmacophore?

80
Functional Group Modification
Antibacterial agent carbutamide (2.64, R = NH2) has an antidiabetic side effect. Replacement of NH2 by CH3 gives tolbutamide (2.64, R = CH3), which is an antidiabetic drug with no antibacterial activity. An obvious relationship exists between molecular structure and its activity.
 has an antidiabetic side effect. Replacement of NH2 by CH3 gives tolbutamide (2.64, R = CH3), which is an antidiabetic drug with no antibacterial activity. An obvious relationship exists between molecular structure and its activity.")
81
Antihypertensives Antihypertensive with diuretic activity
Antihypertensive without diuretic activity

82
Structure-Activity Relationships (SARs)
Crum-Brown and Fraser Examined neuromuscular blocking effects of a variety of simple quaternary ammonium salts to determine if the quaternary amine in curare was the cause for its muscle paralytic properties. Conclusion: the physiological action is a function of chemical constitution

83
Curare alkaloid Figure 2.16 Structure of d-tubocurarine, a constituent of curare

84
Structurally specific drugs (most drugs):
Act at specific sites (receptor or enzyme) Activity/potency susceptible to small changes in structure Structurally nonspecific drugs: No specific site of action Similar activities with varied structures (various gaseous anesthetics, sedatives, antiseptics)
 Activity/potency susceptible to small changes in structure. Structurally nonspecific drugs: No specific site of action. Similar activities with varied structures (various gaseous anesthetics, sedatives, antiseptics)")
85
Example of SAR Lead: sulfanilamide (R = H)
Thousands of analogs synthesized From clinical trials, various analogs shown to possess three different activities: Antimicrobial Diuretic Antidiabetic

86
SAR General Structure of Antimicrobial Agents
R = SO2NHR, SO3H Groups must be para Must be NH2 (or converted to NH2 in vivo) Replacement of benzene ring or added substituents decreases or abolishes activity R can be (but potency is reduced) R = SO2NR2 gives inactive compounds
 Replacement of benzene ring or added substituents decreases or abolishes activity. R can be. (but potency is reduced) R = SO2NR2 gives inactive compounds.")
87
SAR Antidiabetic Agents
X = O, S, or N

88
SAR Diuretics (2 types) hydrochlorothiazides
R2 is an electrophilic group high ceiling type (high level of diuresis)
")
89
SAR for Paclitaxel Paclitaxel (2.72, Taxol) - anticancer drug
 - anticancer drug")
90
SAR Conclusions Use of a molecular activity map-
Figure 2.17 Use of a molecular activity map- structural drawing of a lead annotated to show where structural changes affect activity or potency

91
Structural Modifications
Increase potency, therapeutic index and ADME Increase therapeutic index - measure of the ratio of the concentration of a drug that gives undesirable effects to that which gives desirable effects e.g., LD50 (lethal dose for 50% of the test animals) ED50 (effective dose to give maximum effect in 50% of test animals) Therefore, want LD50 to be large and ED50 to be small. The larger the therapeutic index, the greater the margin of safety. The more life threatening the disease, the lower is an acceptable therapeutic index.
 ED50 (effective dose to give maximum effect in 50% of test animals) Therefore, want LD50 to be large and ED50 to be small. The larger the therapeutic index, the greater the margin of safety. The more life threatening the disease, the lower is an acceptable therapeutic index.")
92
Chlorambucil, an antitumor drug
Therapeutic index = 23

93
Types of Structural Modifications
Homologation - increasing compounds by a constant unit (e.g., CH2) Effect of carbon chain length on drug potency Figure 2.18 Pharmacokinetic explanation: Increasing chain length increases lipophilicity and ability to cross membranes; if too high lipophilicity, it remains in the membrane Pharmacodynamic explanation: Hydrophobic pocket increases binding with increasing length; too large and does not fit into hydrophobic pocket
 Effect of carbon chain length on drug potency. Figure Pharmacokinetic explanation: Increasing chain length increases lipophilicity and ability to cross membranes; if too high lipophilicity, it remains in the membrane. Pharmacodynamic explanation: Hydrophobic pocket increases binding with increasing length; too large and does not fit into hydrophobic pocket.")
94
Branched chain groups are less lipophilic than straight chain groups.
in throat lozenges Branched chain groups are less lipophilic than straight chain groups.

95
More potent than the tertiary homolog
Chain Branching Often lowers potency and/or changes activity; interferes with receptor binding More potent than the tertiary homolog

96
Phenothiazines All bind to different receptors
10-Aminoalkylphenothiazines (X = H) Promethazine-antispasmodic/antihistamine activities predominate Promazine-greatly reduced antispasmodic/antihistamine Activities, greatly enhanced sedative/tranquilizing activities Trimepazine-reduced tranquilizing activity enhanced antipruritic (anti-itch) activity
 Promethazine-antispasmodic/antihistamine. activities predominate. Promazine-greatly reduced antispasmodic/antihistamine. Activities, greatly enhanced sedative/tranquilizing activities. Trimepazine-reduced tranquilizing activity. enhanced antipruritic (anti-itch) activity.")
97
Bioisosterism Bioisosteres - substituents or groups with chemical or physical similarities that produce similar biological properties. Can attenuate toxicity, modify activity of lead, and/or alter pharmacokinetics of lead.

98
Classical Isosteres

99
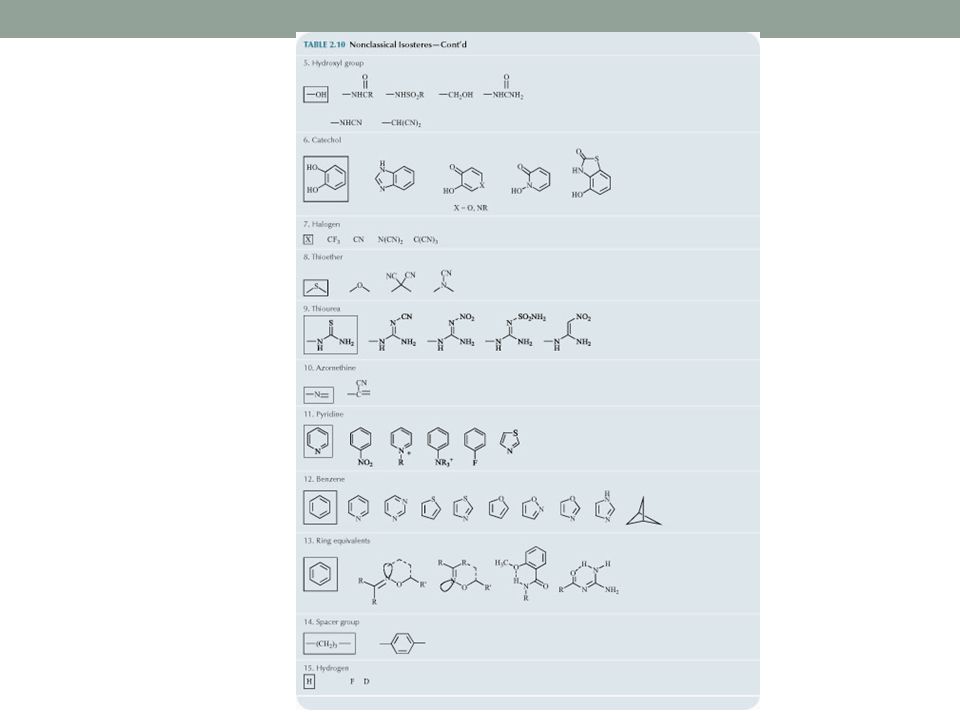
Non-Classical Isosteres
Do not have the same number of atoms and do not fit steric and electronic rules of classical isosteres, but have similar biological activity.

101
Examples of Bioisosteric Analogues

102
Changes in Activity by Bioisosterism
If the S in phenothiazine neuroleptic drugs (2.75) is replaced by -CH=CH- or -CH2-CH2- bioisosteres, then dibenzazepine antidepressant drugs (2.76) result.
 is. replaced by -CH=CH- or -CH2-CH2- bioisosteres, then dibenzazepine antidepressant drugs (2.76) result.")
103
Changes resulting from bioisosteric replacements:
Size, shape, electronic distribution, lipid solubility, water solubility, pKa, chemical reactivity, hydrogen bonding Effects of bioisosteric replacement: 1. Structural (size, shape, H-bonding are important) 2. Receptor interactions (all but lipid/H2O solubility are important) 3. Pharmacokinetics (lipophilicity, hydrophilicity, pKa, H-bonding are important) 4. Metabolism (chemical reactivity is important) Bioisosteric replacements allow you to tinker with whichever parameters are necessary to increase potency or reduce toxicity.
 2. Receptor interactions (all but lipid/H2O solubility are important) 3. Pharmacokinetics (lipophilicity, hydrophilicity, pKa, H-bonding are important) 4. Metabolism (chemical reactivity is important) Bioisosteric replacements allow you to tinker with whichever parameters are necessary to increase potency or reduce toxicity.")
104
Bioisosterism allows modification of physicochemical parameters
Multiple alterations may be necessary: If a bioisosteric modification for receptor binding decreases lipophilicity, you may have to modify a different part of the molecule with a lipophilic group. Where on the molecule do you go to make the modification? The auxophoric groups that do not interfere with binding.

105
Conformationally rigid analogs
Figure 2.19 Conformationally rigid analogs to determine bioactive conformation

106
Ring-Chain Transformations
Transformation of alkyl substituents into cyclic analogs, which generally does not affect potency. Ring-chain transformation can have pharmacokinetic effects, such as increased lipophilicity or decreased metabolism. 2.80 and 2.81 have equivalent tranquilizing effects 2.82. and 2.83 have similar antipruritic activity 2.84 has better antiemetic activity than 2.80

107
Ring-chain transformations can affect toxicity
nontoxic

108
Peptidomimetics rapidly proteolyzed in GI tract and serum
Peptides are important endogenous molecules - neurotransmitters, hormones, neuromodulators Peptide drugs - analgesics, antihypertensives, antitumor agents Peptides generally do not make good drug candidates rapidly proteolyzed in GI tract and serum poorly bioavailable rapidly excreted bind to multiple receptors Peptidomimetic - a compound that mimics or blocks the biological effect of a peptide, but without undesirable structural characteristics

109
Use the peptide as a lead - modify to minimize undesirable pharmacokinetic properties
Try to mimic structure of the peptide when it is bound to the target receptor Replace as much of the peptide backbone as possible with nonpeptide fragments - leave the pharmacophoric groups Initially, retain conformational flexibility, but then refine to more conformationally-rigid analogs to hold groups in a bioactive conformation.

110
Captopril-inhibits angiotensin converting enzyme

111
Phenylalanine Peptidomimetics
Figure 2.20 Conformationally restricted phenylalanine analogs Increased lipophilicity and conformational rigidity - better absorption and poor recognition by proteases

112
Conformationally-Restricted Peptides
Figure 2.21 Conformationally restricted dipeptide analogs

113
Secondary Structure Mimetics
Figure 2.22 Conformationally restricted secondary structure peptidomimetics --turns- -helix -strand -loop

114
Scaffold Peptidomimetics
Arg-Gly-Asp (RGD) common -turn motif that binds to receptors Figure 2.23 RGD scaffold peptidomimetics
 common -turn motif that binds to receptors. Figure 2.23 RGD scaffold peptidomimetics.")
115
somatostatin agonist scaffold peptidomimetic

116
Thyrotropin-releasing hormone (TRH)
A scaffold peptidomimetic for TRH

117
Peptide Backbone Isosteres Peptide amide bond replaced with alternative groups
(statine)
")
118
Pseudopeptides

119
Mechanism of peptide hydrolysis
Figure 2.24 Hydrolysis of a peptide bond showing the tetrahedral intermediate arising from nucleophilic attack of water on the carbonyl group of the peptide bond. The hydroxyethylene isostere analog is designed to mimic the tetrahedral intermediate.

120
Structure Modifications to Increase Oral Bioavailability
~ 75% of drug candidates do not go to clinical trials because of pharmacokinetic problems Less than 10% of drug candidates in clinical trials go to market ~ 40% of molecules that fail in clinical trials do so because of pharmacokinetic problems Therefore, examine pharmacokinetic problems, such as oral bioavailability and plasma half life, as early as possible in the drug discovery process.

121
Low water solubility (high lipophilicity) can be an important limiting factor for oral bioavailability. Highly lipophilic compounds are easily metabolized or bind to plasma proteins. Low lipophilicity leads to poor absorption (cannot cross membranes). Therefore, need to know lipophilicities of molecules and of substituents. Determination of lipophilicities of substituents by Corwin Hansch and co-workers is based on the Hammett equation for electronic effects.
. Therefore, need to know lipophilicities of molecules and of substituents. Determination of lipophilicities of substituents by Corwin Hansch and co-workers is based on the Hammett equation for electronic effects.")
122
Electronic Effects The Hammett Equation
Hammett’s postulate: Electronic effects (both inductive and resonance) of a set of substituents should be similar for different organic reactions. Therefore, assign values for the electronic effect of different substituents in a standard organic reaction, then use these values to estimate rates in a new reaction.
 of a set of substituents should be similar for different organic reactions. Therefore, assign values for the electronic effect of different substituents in a standard organic reaction, then use these values to estimate rates in a new reaction.")
123
Hammett Equation Standard system: benzoic acid derivatives
Equilibrium Constants Scheme 2.7 As X becomes more e--withdrawing, the reaction to the right should be favored (increased Ka).
.")
124
Rate Constants Scheme 2.8 As X becomes more e--withdrawing, rate should increase because of transition state stabilization (lower activation energy).
.")
125
Linear Free-Energy Relationship
Rates of hydrolysis of ethyl benzoates not for ortho substituents- steric/polar effects Dissociation constants of benzoic acids

126
k K log = log ko Ko slope of line ko and Ko are rate and equilibrium constants, respectively, for the parent compound (X = H) K If is defined as , log Ko k then log = Hammett equation ko Electronic parameter (depends on electronic properties of substituent) the more e--withdrawing, more + the more e--donating, more - Reaction constant (depends on reaction conditions) carbocation intermediate, - slope carbanion intermediate, + slope H = 0.0
 the more e--withdrawing, more + the more e--donating, more - Reaction constant (depends on reaction conditions) carbocation intermediate, - slope. carbanion intermediate, + slope. H = 0.0.")
127
Lipophilicity Effects: Hansch Equation
Hansch thought there should be a linear free-energy relationship between lipophilicity and biological activity. Action of drug depends on 2 processes: 1. drug has to get to site of action (pharmacokinetics) 2. drug has to interact with site of action (pharmacodynamics)
 2. drug has to interact with site of action (pharmacodynamics)")
128
Fluid Mosaic Model of Membrane Drug must pass through various membranes to reach the site of action
Figure 2.26 lipid bilayer hydrophilic heads (OH, NH3+, sugars) Correlations noted earlier between lipid solubility and biological activity lipophilic tails (steroid and hydrocarbon chains)
 Correlations noted earlier between lipid solubility and biological activity. lipophilic tails (steroid and hydrocarbon chains)")
129
Lipids found in membranes

130
Measured Lipophilicities Model for transport of drug to site of action
Ability of compound to partition between 1-octanol (simulates membrane) and water (simulates cytoplasm) Measure of lipophilicity: partition coefficient (P) between 1-octanol and water: [compound]1-octanol P = (Eq. 2.7) [compound]water (1-) degree of dissociation in H2O from ionization constants
 and water (simulates cytoplasm) Measure of lipophilicity: partition coefficient (P) between 1-octanol and water: [compound]1-octanol. P = (Eq. 2.7) [compound]water (1-) degree of dissociation in H2O from ionization constants.")
131
Relative potency of drug (log )
1 Relative potency of drug (log ) C concentration of drug that produces the biological effect 1 log = -k(log P)2 + k(log P) + k C For a compound more soluble in H2O than in 1-octanol P < 1; log P is - For a compound more soluble in 1-octanol than in H2O P > 1; log P is + The larger the P, the more interaction of the drug with membranes (more lipophilic)
 C. concentration of drug that produces the biological effect. 1. log = -k(log P)2 + k(log P) + k C. For a compound more soluble in H2O than in 1-octanol. P < 1; log P is - For a compound more soluble in 1-octanol than in H2O. P > 1; log P is + The larger the P, the more interaction of the drug with membranes (more lipophilic)")
132
Parabolic Relationship Between Potency (log ) and log P
1 C optimum partition coefficient for biological activity log P0 should be 2 to penentrate the CNS Figure 2.27 Effect of log P on biological response. P is the partition coefficient, and C is the oncentration of the compound required to produce a standard biological effect. Log Po is the optimal log P for biological activity.

133
Ionization of compounds leads to greater water solubility than predicted from the neutral structure.
For compounds that can ionize (COO-, NH3+, etc.) use log D - log of distribution coefficient. Ionization is a function of pKa of a compound and pH of the medium; therefore, must specify pH. log D is the log P of an ionizable compound at a particular pH. log D7.4 is the log P at pH 7.4
 use log D - log of distribution coefficient. Ionization is a function of pKa of a compound and pH of the medium; therefore, must specify pH. log D is the log P of an ionizable compound at a particular pH. log D7.4 is the log P at pH 7.4.")
134
Effect of pH on log D fully protonated amine
decreased concentration of protonated amine

135
Lipophilicity Substituent Constants p
Derived the same as electronic substituent constants = log PX - log PH = log PX PH log P for a compound with substituent X log P for a compound with no substituent

136
(like ) is additive and constitutive
Multiple substituents exert lipophilicity equal to the sum of individual substituents Effect depends on the molecule to which it is attached Alkyl groups are least constitutive CH3 ≈ 0.50 By definition, H = 0 Therefore, CH2 = CH3

139
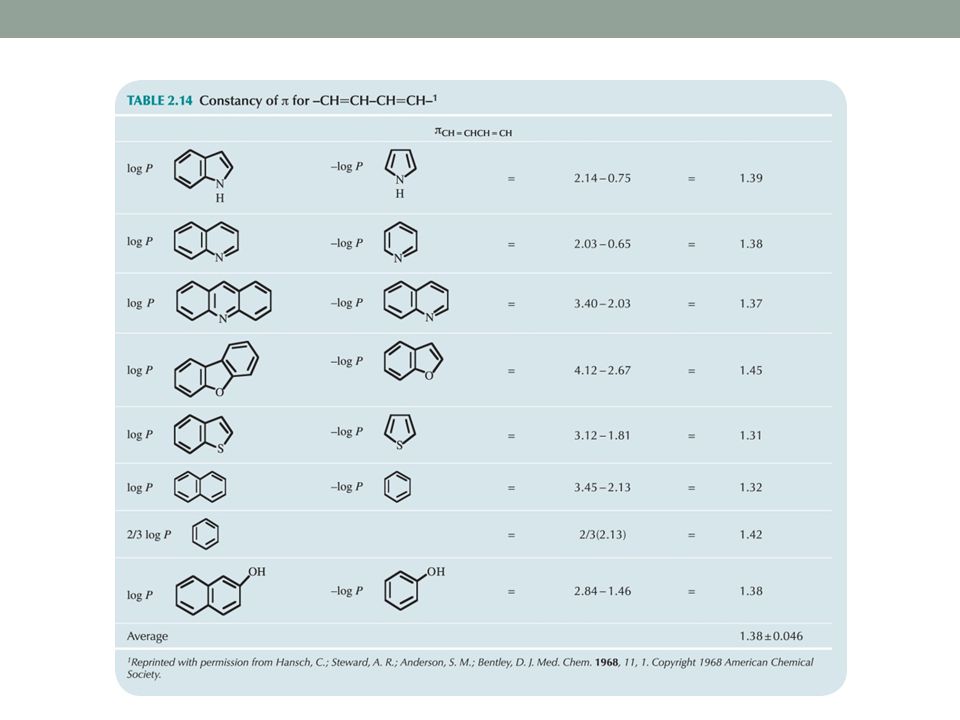
How does folding affect π values?

140
Log P = 2πCH3 + 2πCH2 + πCH=CH + 2logPPhOH-0.40
= 2(0.50)+2(0.50) (1.46)-0.40 =5.21 (experimental, 5.07)
+2(0.50) (1.46) =5.21 (experimental, 5.07)")
141
Example of Additivity of Constants Branching lowers log P or by 0
Example of Additivity of Constants Branching lowers log P or by 0.2/branch branching log P2.83 = 2Ph + CH + OCH2 + CH2 + NMe OCH2 = log PCH3CH2OCH2CH3 - 2CH3 - CH2 = (0.5) = -0.73 NMe2 = log PPh(CH2)3NMe2 - Ph - 3CH2 = (0.5) = -0.95 log P2.83 = 2(2.13) (-0.73) (-0.95) - 0.2 = 3.38 branching Experimental log P = 3.27
 = NMe2 = log PPh(CH2)3NMe2 - Ph - 3CH2. = (0.5) = log P2.83 = 2(2.13) (-0.73) (-0.95) = branching. Experimental log P =")
142
Computerization of Log P Values
Several software packages are available to calculate log P. However, log P values can vary by 2 or more units among software packages. values are constitutive - cannot account for all substituents on every scaffold. Measure log P for one member of a library, then see which software package comes closest to the experimental value.

143
Effects of Ionization on Lipophilicity and Oral Bioavailability
Amino acid residues in receptors are ionized Asp/Glu -COOH pKa Cys -SH Tyr Ser/Thr -OH His Lys -NH Arg At physiological pH (pH 7.4) each is partially ionized and partially neutral. pH = pKa –log [HA]/[A-]
 each is partially ionized and partially neutral. pH = pKa –log [HA]/[A-]")
144
Ionization of a drug depends on pKa of groups and pH.
Ionization has profound effect on lipophilicity (pharmacokinetics) and interaction with a receptor (pharmacodynamics). What if ionization of a drug is important for its binding to a receptor, but ionization may block its ability to cross membranes? Need to vary pKa to adjust equilibrium of ionized (bind to receptor) and neutral (cross membranes) form. Neutral form crosses membranes, then re-establishes equilibrium with ionized form on other side.
 and interaction with a receptor (pharmacodynamics). What if ionization of a drug is important for its binding to a receptor, but ionization may block its ability to cross membranes Need to vary pKa to adjust equilibrium of ionized (bind to receptor) and neutral (cross membranes) form. Neutral form crosses membranes, then re-establishes equilibrium with ionized form on other side.")
145
Ionized molecules that did not cross the membrane re-establish equilibrium with the neutral form, which can cross the membrane. Drug metabolism and excretion prevent continual re-establishment of equilibrium. The pKa of most alkaloids that act as neuroleptics, local anesthetics, and barbituates have values between 6 and 8. At neutral pH there is a mixture of neutral and cationic forms.

146
Aminacrine is more active at pH > 7

147
Sulfinpyrazone (R=CH2CH2SOCH3)has a pKa of 2.8
The uricosuric drug phenylbutazone has a pKa of 4.5 and is active as an anion. Scheme 2.10 Ionization equilibrium for phenylbutazone The pH of urine is ~ 4.8. Sulfinpyrazone (R=CH2CH2SOCH3)has a pKa of 2.8 Therefore all in anionic form. 20 times more potent than phenylbutazone
has a pKa of 2.8. Therefore all in anionic form. 20 times more potent than phenylbutazone.")
148
Effect of ionization can be rationalized from pharmacokinetic or pharmacodynamic perspective.
Pharmacokinetic - neutral form increases crossing of membranes Pharmacodynamic - neutral form binds in a hydrophobic pocket in receptor These can be differentiated using an isolated receptor (no membranes to cross so only pharmacodynamics) and whole cells or tissue (membranes to cross so pharmacokinetics and pharmacodynamics are important).
 and whole cells or tissue (membranes to cross so pharmacokinetics and pharmacodynamics are important).")
149
Pyrimethamine is absorbed as the neutral
molecule, but active as the protonated ion

150
In a cell-free system (no membranes), the antibacterial activity of sulfamethoxazole is directly proportional to the degree of ionization (pharmacodynamics). Scheme 2.11 Ionization equilibrium for sulfamethoxazole In intact cells, where a drug must cross a membrane to get to the site of action, the antibacterial activity is proportional to its lipophilicity (neutral).
.")
151
Actual pKa Values pKa values depend on the microenvironment
Molecular dynamics simulations indicate that the interior of proteins have dielectric constants about 2-3 (like benzene or dioxane); water has a high dielectric constant (78.5). COOH in a nonpolar environment - pKa higher because ionized form is destabilized. Asp-99 in 3-oxo-5-steroid isomerase is 9.5! Represents a change in equilibrium of 105 in favor of neutral form.
; water has a high dielectric constant (78.5). COOH in a nonpolar environment - pKa higher because ionized form is destabilized. Asp-99 in 3-oxo-5-steroid isomerase is 9.5! Represents a change in equilibrium of 105 in favor of neutral form.")
152
COOH that forms a salt bridge - pKa lower
Multiple COOH groups adjacent - pKa higher to avoid adjacent anionic groups NH2 in a nonpolar environment - pKa lower to avoid cationic form NH2 in a salt bridge is stabilized - pKa higher Multiple basic groups adjacent - pKa lower to avoid adjacent cationic groups Lys amino group in acetoacetate decarboxylase has pKa 5.9 because adjacent residue is another Lys. Again about 105 change in equilibrium.

153
Such large changes in pKa in microenvironment indicate that lead modification also should involve large pKa variances. Determine in vitro and in vivo effects with pKa changes.

155
CNS drugs must cross the blood-brain barrier
Inactive Active Active

156
Quantitative Structure-Activity Relationships
Crum-Brown and Fraser predict a mathematical relationship would be found between structure and activity Corwin Hansch attempts to quantify effects of substituent modifications

157
QSAR Basis for quantitative drug design:
Biological properties are a function of the physicochemical parameters, e.g., solubility, lipophilicity, electronic effects, ionization, stereochemistry, etc. and constants should be important considerations in lead modification. Steric factors also should be important for receptor binding.

158
Steric Effects: Taft Equation
Taft defined a steric parameter Es Reference reaction: XCH2CO2Me XCH2CO2H + MeOH H3O+ Es (X = H) = 0 CH3 k0 Es = log kXCH2Me - log kCH3CO2Me = log kX Hancock: Ecs= Es =0.306(n-3) corrects for hyperconjugation
 = 0. CH3. k0. Es = log kXCH2Me - log kCH3CO2Me = log. kX. Hancock: Ecs= Es =0.306(n-3) corrects for hyperconjugation.")
159
regression coefficients potency Hansch Equation
Correlation of Physicochemical Parameters (descriptors) with Biological Activity Hansch analysis Because there are at least two considerations for drug activity, lipophilicity (to get drug to the site of action) and electronic factors (at the site of action), Hansch derived: 1 log = -k2 + k + + k C regression coefficients potency Hansch Equation
 with Biological Activity. Hansch analysis. Because there are at least two considerations for drug activity, lipophilicity (to get drug to the site of action) and electronic factors (at the site of action), Hansch derived: 1. log = -k2 + k + + k C. regression coefficients. potency. Hansch Equation.")
160
Hansch equation generalized:
1 log = -a2 + b + + cEs + dS + e C lipophilicity constant electronic constant steric constant other physico-chem. parameters potency Linear multiple regression analysis The best least-squares fit of the dependent variable (biological activity) to a linear combination of independent variables (descriptors) is determined.
 to a linear combination of independent variables (descriptors) is determined.")
161
Topliss Operational Schemes - Lead Optimization
Nonmathematical, nonstatistical, noncomputerized use of Hansch principles. Consider benzenesulfonamide as lead (R = H) Vary and and determine effect on potency. This method requires an unfused benzene ring.
 Vary and and determine effect on potency. This method requires an unfused benzene ring.")
162
Topliss Decision Tree Because potency often increases with lipophilicity, first try a substituent with a + value (e.g., Cl; 4-Cl = 0.71, 4-Cl = 0.23). Three possible outcomes - more potent (M), equipotent (E), or less potent (L) than parent. If more potent, can be because of +, +, or both. Hold one constant and vary other: 4-PhS = 2.32, 4-PhS = 0.18 would test lipophilicity 4-CF3 = 0.88, 4- CF3 = 0.54 would test electron withdrawal If 4-PhS compound is more potent than 4-Cl, increase lipophilicity more.
. Three possible outcomes - more potent (M), equipotent (E), or less potent (L) than parent. If more potent, can be because of +, +, or both. Hold one constant and vary other: 4-PhS = 2.32, 4-PhS = 0.18 would test lipophilicity. 4-CF3 = 0.88, 4- CF3 = 0.54 would test electron withdrawal. If 4-PhS compound is more potent than 4-Cl, increase lipophilicity more.")
163
Topliss Decision Tree Figure 2.16

164
If 4-Cl analog is equipotent with parent, may be a counterbalance of favorable + and unfavorable + or vice versa. Try 4-Me 4-Me = 0.56, 4-Me = or 4-NO2 4- NO2 = -0.28, 4- NO2 = 0.78 If 4-Cl analog is less potent than parent, there may be a steric problem at C-4 or need - and/or -. Try 3-Cl 3-Cl = 0.71, 3-Cl = 0.37

165
Craig plot of σ and π values
Figure 2.29 Craig plot of σ constants versus π values for aromatic substituents. This material is reproduced with the permission of John Wiley & Sons, Inc. From Craig, P. N. (1980) In "Burger’s Medicinal Chemistry," (M. E. Wolff, ed.), 4th ed., Part I, p Wiley, New York.
 In Burger’s Medicinal Chemistry, (M. E. Wolff, ed.), 4th ed., Part I, p Wiley, New York.")
166
Batch Selection Methods
With Topliss operational scheme you need results from one compound before you know what to make next. Batchwise methods involve making a small group of preselected analogs and testing. Rank order the potencies, then compare with the rankings in Table 2.17. This table tells you which parameter is most important. E.g., if rank is 3,4-Cl2 > 4-Cl > H > 4-Me > 4-OMe, then is most important.

167
Then consult Table 2.18 for other substituents with that parameter.
Cannot extend this method without computation.

168
Cluster Analysis Select one member from each cluster
See which dominates

169
Donepezil was studied by QSAR

170
Free-Wilson approach to QSAR
BA = ΣaiXi + μ ai = contribution of a to BA Xi=1 or 0 μ = activity of parent skeleton R1 = R2 = R3 = R4 If R5 is Cl or CH3 and Cl > CH3 Then assume Cl always better, regardless of other substituents

171
Scaffold hopping Figure 2.30 Example of scaffold hopping to identify new cholesterol-lowering statins from mevastatin

172
Scaffold hopping Figure 2.31 A variation of scaffold hopping that involves disconnection of bonds, rotation or flipping of the core, and reconnection of the pharmacophoric groups

173
Computer-Assisted Drug Design Discovery of the influenza drug zanamivir (RelenzaTM)
A protein (hemagglutinin) at the surface of a virus binds to sialic acid residues on receptors at the host cell surface. Sialic acid Lead The virus has another protein (neuraminidase), which is an enzyme that cleaves sialic acid residues from the cell surface The virus enters the cell and replicates in the nucleus.
 at the surface of a virus binds to sialic acid residues on receptors at the host cell surface. Sialic acid. Lead. The virus has another protein (neuraminidase), which is an enzyme. that cleaves sialic acid residues from the cell surface. The virus enters the cell and replicates in the nucleus.")
174
Progeny virus particles escape the cell, which no longer has sialic acid residues on its surface to trap the virus. The virus particles spread to other cells. Inhibition of neuraminidase would prevent the release and spread of virus particles.

175
Random screen identified 2.129 (R = OH) as a weak inhibitor.
Crystal structure of influenza A neuraminidase with inhibitor bound was obtained. NH3+ group Computation (using GRID program) suggested 4-OH should be replaced by 4-NH2, which when protonated would interact with Glu-119 (Fig 2.32). From the crystal structure, extension of 4-ammonium with 4-guanidinium (2.93, zanamivir) would extend binding to Glu-227 as well (Fig. 2.32). group Figure 2.32 Crystal structure of neuraminidase active site with inhibitors bound. (A) Interaction of the protonated amino group of (R = NH3+) with Glu-119. (B) Interaction of the protonated guanidinium group of with Glu-119 and (red arrow) Glu-227. Adapted with permission from Macmillan Publishers Ltd: Nature (von Itzstein, M., et al. Rational design of potent sialidase-based inhibitors of influence virus replication. Nature 1993, 363, 418–423) Copyright 1993.
 suggested 4-OH should be replaced by 4-NH2, which when protonated would interact with Glu-119 (Fig 2.32). From the crystal structure, extension of 4-ammonium with 4-guanidinium (2.93, zanamivir) would extend binding to Glu-227 as well (Fig. 2.32). group. Figure 2.32 Crystal structure of neuraminidase active site with inhibitors bound. (A) Interaction of the protonated amino group of (R = NH3+) with Glu-119. (B) Interaction of the protonated guanidinium group of with Glu-119 and (red arrow) Glu-227. Adapted with permission from Macmillan Publishers Ltd: Nature (von Itzstein, M., et al. Rational design of potent sialidase-based inhibitors of influence virus replication. Nature 1993, 363, 418–423) Copyright")
176
Generally, drug discovery is not this easy - need a cyclic approach.
Identify receptor binder Obtain crystal structure of receptor with molecule bound Do calculations and docking Synthesize new compounds Assay Obtain another crystal structure with more potent binder Repeat cycle

177
Computer modeling can be used to identify the pharmacophore.
By overlaying the structure of the antitumor drug paclitaxel (2.72, Taxol) with those of 4 other natural products that also stabilize microtubules in competition with paclitaxel, (see next slide)
 with those of 4 other. natural products that also stabilize microtubules in competition with paclitaxel, (see next slide)")
178
a common pharmacophore (Fig. 2.34, next slide) was identified.
Figure 2.33 Five natural products found to promote stabilization of microtubules. The boxed sections were used to identify a common pharmacophore. With permission for I. Ojima (1999). Reprinted with permission from Proc. Natl. Acad. Sci. USA 1999, 96, 4256–4261. Copyright 1999 National Academy of Sciences, U.S.A. a common pharmacophore (Fig. 2.34, next slide) was identified.
. Reprinted with permission from Proc. Natl. Acad. Sci. USA 1999, 96, 4256–4261. Copyright 1999 National Academy of Sciences, U.S.A. a common pharmacophore (Fig. 2.34, next slide) was identified.")
179
A hybrid structure was then constructed (2.130).
Figure 2.34 Common pharmacophore based on the composite of boxed sections in Figure With permission for I. Ojima (1999). Reprinted with permission from Proc. Natl. Acad. Sci. USA 1999, 96, 4256–4261. Copyright 1999 National Academy of Sciences, U.S.A.
. Reprinted with permission from Proc. Natl. Acad. Sci. USA 1999, 96, 4256–4261. Copyright 1999 National Academy of Sciences, U.S.A.")
180
The design of a bryostatin analogue

Similar presentations










 Comparative Molecular Field Analysis (CoMFA) Gijs Schaftenaar.>")

 and Comparative Molecular Field Analysis (CoMFA) Martin Ott.>")





 Attempts to identify and quantitate physicochemical properties of a drug in relation to its biological.>")



