Download presentation
Presentation is loading. Please wait.

1
WP6 Part 1: Bioinformatics First results passed peer review Working on more extensive proteomics knowledge sharing Library of existing services collated Library of LCC experiment protocols underway Presenters: Xueping Quan, Marco Schorlemmer, Dave Robertson

2
OK From an Experimenter’s Viewpoint Interaction model = Experiment design – Experimental roles allocated to peers – Constraints prescribe methods on peers – Message passing synchronises tasks Formal model gives: –Automation, extending experiment repertoire –Repeatability, because we preserve state –Scrutiny, for reviewers

3
P2P Proteomics Proteome is the protein equivalent of the genome Proteomics studies the quantitative changes occurring in a proteome and its application for –disease diagnostics –therapy –drug development

4
Peer-to-Peer Experimentation in Protein Structure Prediction: an Architecture, Experiment and Initial Results

5
Experiment - Consistency Checking Taking a non-expert user’s perspective… Applied Bioinformatics - Whom to believe?? Note: This Scenario needs to allow for “passive” peers to incorporate knowledge from the large number of traditional bioinformatics resources (databases etc.) Comparison of server results for consistency typically increases confidence in the result.
 Comparison of server results for consistency typically increases confidence in the result..")
6
Experiment – “Consistency Checking” Step1: Proxy per service allowing data retrieving from “passive” peers. Each query is related to the appropriate service. query (input, keyword, ID, sequence, etc. ) data relating to input Proxies (Wrappers) Interfaces (WSDL, etc) ApplicationDatabaseWeb Server
 data relating to input Proxies (Wrappers) Interfaces (WSDL, etc) ApplicationDatabaseWeb Server.")
7
Experiment – “Consistency Checking” Local database of trusted results with provenance Polling multiple sites Step 2: Automated harvesting of results for targets and collation to allow easy comparison of answers. Scientist logs local opinion on relative quality of (passive) other peers for each target and caches the most important positive and/or negative results.
 other peers for each target and caches the most important positive and/or negative results..")
8
Extend structural knowledge through modelling: Find fragments of 3D-models of S.cerevisiae (yeast) proteins that can be trusted 6604 yeast protein sequences (some predicted) currently 330 known 3D-structures (in PDB) Experiment: Specific Task (Popular strategy, typically accomplished with the help of a meta-WWW-server today.)
 proteins that can be trusted 6604 yeast protein sequences (some predicted) currently 330 known 3D-structures (in PDB) Experiment: Specific Task (Popular strategy, typically accomplished with the help of a meta-WWW-server today.)")
9
Databases of pre-computed 3D-models SWISS restrictive + non-redundant high-quality models only (SWISSMODEL) SAM yeast models “complete” (at least one model per ID) + redundant; raw models (SAM-T06 / UNDERTAKER) ModBase permissive + highly redundant pre-filtered before the task (PSI-BLAST / MODELLER)
 SAM yeast models complete (at least one model per ID) + redundant; raw models (SAM-T06 / UNDERTAKER) ModBase permissive + highly redundant pre-filtered before the task (PSI-BLAST / MODELLER)")
10
Complications – True and False Redundancy Example 1: highly redundant set Example 2: multi-domain proteins “ non-redundant ” sets (< 90% overlap)
")
11
Databases of pre-computed 3D-models SWISS 769 models SAM yeast models 2211 models (selected top model if E-value < 10 -3 ) ModBase 2546 models (pre-filtered: sequence-id > 20% score > 0.7 E-value < 10 -6 )
 ModBase 2546 models (pre-filtered: sequence-id > 20% score > 0.7 E-value < )")
12
multi-agent interaction coordination through service composition LCC interpreter loosely based on electronic societies (of peers) uses WSDL as standard For more information please refer to: Xueping Quan, Chris Walton, Dietlind L Gerloff, Joanna L Sharman and Dave Robertson, GCCB2006. to be superseded by (more flexible) OK-kernel Implementation using LCC interpreter
 OK-kernel Implementation using LCC interpreter.")
13
Implementation using LCC Interpreter Storing “good answers” in local database ModBase(filtered) SAM SWISS SWISS Service SAM Service ModBase Service LCC Interpreter CYSP Service CYSP MaxSub MaxSub Service HTML WSDL Pair-wise comparison of 3D-protein models
 SAM SWISS SWISS Service SAM Service ModBase Service LCC Interpreter CYSP Service CYSP MaxSub MaxSub Service HTML WSDL Pair-wise comparison of 3D-protein models")
14
a(data_collator, X):: data_request(Is) <= a(experimenter, E) then a(data_collector(Is,Sp,Sd),X) yeast_id(Is) and source(Sp) then filter(Is,Sp,Sd) => a(data_filter((Is,Sp,Sd),F) then filtered(Is,Sp,S) <= a(data_filter(Is,Sp,Sd),F) then filtered(Is,Sp,S) => a(data_comparer,C) then data_compared(Is,SF) <= a(data_comparer,C) then data_compared(Is,SF) => a(experimenter,E) then data_compared(Is,SF) => a(data_publisher,PU) a(experimenter, E):: data_request(Is) => a(data_collator, X) then data_compared(Is,SF) <= a(data_collator, X) a(data_collector(Is,Sp,Sd),X):: ( null Sp=[] and Sd=[]) or ( a(data_retriever(I,P,D),X) (Sp=[P|Rp] and Sd=[D|Rd] and Is=[I|Ri]) then a(data_collector(Ri,Rp,Rd),X) ) a(data_retriever(I,P,D),X):: data_request(I) => a(data_source,P) then data_report(I,D) <= a(data_source,P) a(data_filter(I,Sp,Sd),F):: filter(I,Sp,Sd) <= a(data_collator,X) then filtered(I,Sp,S) => a(data_collator,X) apply_filter(Sd,S) a(data_source,P):: data_request(I) <= a(data_retriever(I,P,D),X) then data_report(I,D) => a(data_retriever(I,P,D),X) lookup(I,D) a(data_comparer,C):: filtered(Is,Sp,S) <= a(data_collator,X) then data_compared(Is,SF) => a(data_collator,X) consistency_check(S,SF) LCC Protocol
![a(data_collator, X):: data_request(Is) <= a(experimenter, E) then a(data_collector(Is,Sp,Sd),X) yeast_id(Is) and source(Sp) then filter(Is,Sp,Sd) => a(data_filter((Is,Sp,Sd),F) then filtered(Is,Sp,S) <= a(data_filter(Is,Sp,Sd),F) then filtered(Is,Sp,S) => a(data_comparer,C) then data_compared(Is,SF) <= a(data_comparer,C) then data_compared(Is,SF) => a(experimenter,E) then data_compared(Is,SF) => a(data_publisher,PU) a(experimenter, E):: data_request(Is) => a(data_collator, X) then data_compared(Is,SF) <= a(data_collator, X) a(data_collector(Is,Sp,Sd),X):: ( null Sp=[] and Sd=[]) or ( a(data_retriever(I,P,D),X) (Sp=[P|Rp] and Sd=[D|Rd] and Is=[I|Ri]) then a(data_collector(Ri,Rp,Rd),X) ) a(data_retriever(I,P,D),X):: data_request(I) => a(data_source,P) then data_report(I,D) <= a(data_source,P) a(data_filter(I,Sp,Sd),F):: filter(I,Sp,Sd) <= a(data_collator,X) then filtered(I,Sp,S) => a(data_collator,X) apply_filter(Sd,S) a(data_source,P):: data_request(I) <= a(data_retriever(I,P,D),X) then data_report(I,D) => a(data_retriever(I,P,D),X) lookup(I,D) a(data_comparer,C):: filtered(Is,Sp,S) <= a(data_collator,X) then data_compared(Is,SF) => a(data_collator,X) consistency_check(S,SF) LCC Protocol](http://images.slideplayer.com/8/2427514/slides/slide_14.jpg "a(data_collator, X):: data_request(Is) <= a(experimenter, E) then a(data_collector(Is,Sp,Sd),X) yeast_id(Is) and source(Sp) then filter(Is,Sp,Sd) => a(data_filter((Is,Sp,Sd),F) then filtered(Is,Sp,S) <= a(data_filter(Is,Sp,Sd),F) then filtered(Is,Sp,S) => a(data_comparer,C) then data_compared(Is,SF) <= a(data_comparer,C) then data_compared(Is,SF) => a(experimenter,E) then data_compared(Is,SF) => a(data_publisher,PU) a(experimenter, E):: data_request(Is) => a(data_collator, X) then data_compared(Is,SF) <= a(data_collator, X) a(data_collector(Is,Sp,Sd),X):: ( null Sp=[] and Sd=[]) or ( a(data_retriever(I,P,D),X) (Sp=[P|Rp] and Sd=[D|Rd] and Is=[I|Ri]) then a(data_collector(Ri,Rp,Rd),X) ) a(data_retriever(I,P,D),X):: data_request(I) => a(data_source,P) then data_report(I,D) <= a(data_source,P) a(data_filter(I,Sp,Sd),F):: filter(I,Sp,Sd) <= a(data_collator,X) then filtered(I,Sp,S) => a(data_collator,X) apply_filter(Sd,S) a(data_source,P):: data_request(I) <= a(data_retriever(I,P,D),X) then data_report(I,D) => a(data_retriever(I,P,D),X) lookup(I,D) a(data_comparer,C):: filtered(Is,Sp,S) <= a(data_collator,X) then data_compared(Is,SF) => a(data_collator,X) consistency_check(S,SF) LCC Protocol")
15
MaxSub - Examples SWISS-SAM ModBase-SAM SWISS-ModBase YPL132W YBR024W YLR131C pair-wise, sequence- dependent finds common substructure (shown in blue)
")
16
CYSP = Comparison of Yeast 3D Structure Predictions 578 three-way supported MaxSub-substructures > 45 aa from 545 proteins (Linked from www.openk.org ) Pair-wise MaxSub Comparisons: Results SWISSModBaseSAM SWISS769 (717)649 (594)585 (559) ModBase2546 (2280)620 (594) SAM2211 (2211)
 Pair-wise MaxSub Comparisons: Results SWISSModBaseSAM SWISS769 (717)649 (594)585 (559) ModBase2546 (2280)620 (594) SAM2211 (2211)")
17
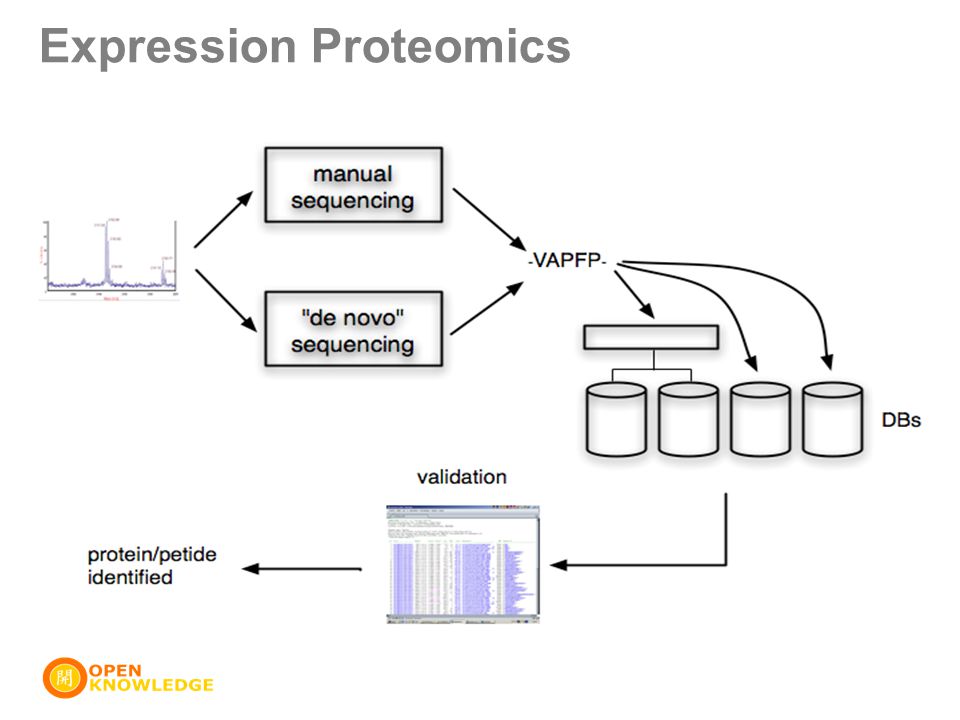
Proteomic Analysis Expression Proteomics –proteins are extracted from cells and tissues –proteins are separated two dimensional cell electrophoresis liquid chromatography –proteins are digested and identified various mass spectrometry methods Bioinformatic Analysis –primary, secondary, tertiary structures –sequence alignment and homology –motifs and domains –protein interactions and networks Functional Proteomics

18
Expression Proteomics

20
Peptide/Protein Identification Sequencing information in archives that do not produce clear identifications rarely accessible to other groups –most part of it will never be reflected in protein DBs –information is trashed Information of high importance for other groups analysing sequence/function of homologue proteins –contains sequences with post-translational modifications not to be found in current protein DBs Spectra and sequence tags generated in one lab could be used by other labs to evaluate confidence of experimental or predicted sequences

21
Information Overflow Proteomic analysis is currently an inhumane task: –LC-MS analysis produces >10,000 of spectra –each spectra yields (after sequencing and DB search) several peptide or peptide tag candidates –each step produces an identification score whose final evaluation is performed manually (using probability data) Many proteomic labs are involved in the characterization of proteomes, protein complexes and networks speed of information production increases very fast
 several peptide or peptide tag candidates –each step produces an identification score whose final evaluation is performed manually (using probability data) Many proteomic labs are involved in the characterization of proteomes, protein complexes and networks speed of information production increases very fast")
22
Expression Proteomics

23
P2P Proteomics with OK

24
Sequence Identification Scenario An investigator asks an identifier to match a sequence against proteomic labs repositories. The identifier acts as a searcher inquiring each known proteomics lab retrieving hits for the given input sequence, collects results, and then sends them back to investigator. The inquired proteomics lab could store high scoring queries to increase the reliability of the matching sequences. The end-point process of sequence data-mining done by the proteomics lab is performed by Blast engines local to each peer. The first prototype only matches input sequences; next release could also directly accept mass spectra as input. For this task will us an OMSSA engine capable of matching spectra against the same sequence database used by Blast engine.

25
Sequence Identification IM in LCC a(investigator,A) :: identify(Seqs,P) => a(identifier,B) get_sequences(Seqs,P) then visualise(Result_set) answer(Result_set) <= a(identifier,B) a(identifier,B) :: identify(Seqs,P) <= a(investigator,A) then a(searcher(Seqs,P,Ls,Result_set),B) lab_list(Ls) then answer(Result_set) => a(investigator,A) then a(identifier,B) a(searcher(Seqs,P,Ls,Result_set),B) :: ( query(Seqs,P) => a(proteomics_lab,L) Ls = [L|RLs] then Result_set = [(Result,L)|RSs] answer(Result) <= a(proteomics_lab,L) then a(searcher(Seqs,P,RLs,RSs) ) or null Ls = [] and Result_set = [] a(proteomics_lab,L) :: query(Seqs,P) <= a(searcher(_,_,_,_),B) then answer(Result) => a(searcher(_,_,_,_),B) find_hit(Seqs,P,Result) then a(proteomics_lab,L)
![Sequence Identification IM in LCC a(investigator,A) :: identify(Seqs,P) => a(identifier,B) get_sequences(Seqs,P) then visualise(Result_set) answer(Result_set) <= a(identifier,B) a(identifier,B) :: identify(Seqs,P) <= a(investigator,A) then a(searcher(Seqs,P,Ls,Result_set),B) lab_list(Ls) then answer(Result_set) => a(investigator,A) then a(identifier,B) a(searcher(Seqs,P,Ls,Result_set),B) :: ( query(Seqs,P) => a(proteomics_lab,L) Ls = [L|RLs] then Result_set = [(Result,L)|RSs] answer(Result) <= a(proteomics_lab,L) then a(searcher(Seqs,P,RLs,RSs) ) or null Ls = [] and Result_set = [] a(proteomics_lab,L) :: query(Seqs,P) <= a(searcher(_,_,_,_),B) then answer(Result) => a(searcher(_,_,_,_),B) find_hit(Seqs,P,Result) then a(proteomics_lab,L)](http://images.slideplayer.com/8/2427514/slides/slide_25.jpg "Sequence Identification IM in LCC a(investigator,A) :: identify(Seqs,P) => a(identifier,B) get_sequences(Seqs,P) then visualise(Result_set) answer(Result_set) <= a(identifier,B) a(identifier,B) :: identify(Seqs,P) <= a(investigator,A) then a(searcher(Seqs,P,Ls,Result_set),B) lab_list(Ls) then answer(Result_set) => a(investigator,A) then a(identifier,B) a(searcher(Seqs,P,Ls,Result_set),B) :: ( query(Seqs,P) => a(proteomics_lab,L) Ls = [L|RLs] then Result_set = [(Result,L)|RSs] answer(Result) <= a(proteomics_lab,L) then a(searcher(Seqs,P,RLs,RSs) ) or null Ls = [] and Result_set = [] a(proteomics_lab,L) :: query(Seqs,P) <= a(searcher(_,_,_,_),B) then answer(Result) => a(searcher(_,_,_,_),B) find_hit(Seqs,P,Result) then a(proteomics_lab,L)")
26
investigator get_sequence (Seqs, P) GUI identifier identify(Seqs, P) answer(result_set) searcher query(Seqs, P) answer(result) identifier investigator visualise (result_set) GUI proteomics_lab An investigator uses a GUI to get an input sequences and a set of parameters P Investigator sends message identify(Seqs, P) to an identifier identifier retrieves a list of known proteomics labs identifier becomes searcher and sends a query to the first proteomics_lab of the list proteomics_lab resolves find_hit constraint and sends back an answer with the result (i.e. an URL for a XML file) searcher loops the queries over the list of proteomics_labs and collects results in a result_set searcher comes back to role identifier and sends back result_set to investigator investigator receives the result_set and displays it on a GUI Step by Step peer message constraint lab_list(Ls) find_hit (Seqs, P) find_hit() constraint also kicks up a process inside proteomics_lab peer which will store high scoring queries
 searcher loops the queries over the list of proteomics_labs and collects results in a result_set searcher comes back to role identifier and sends back result_set to investigator investigator receives the result_set and displays it on a GUI Step by Step peer message constraint lab_list(Ls) find_hit (Seqs, P) find_hit() constraint also kicks up a process inside proteomics_lab peer which will store high scoring queries.")
Similar presentations
 proteome: complete.>")














 Course Director David Fenyö Contact information>")



 Course Director David Fenyö Contact information>")